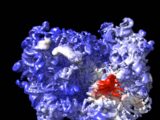
DeepMind’s artificial intelligence predicts the structures of a large number of proteins.
July 23, 2021Over 20,000 proteins are encoded in the human genome. However, approximately one-third of those have had their three-dimensional structures determined experimentally. And in many cases, those structures are unknown in their entirety.
Now, a game-changing artificial intelligence (AI) tool called AlphaFold has predicted the structure of nearly the entire human proteome. AlphaFold was developed by Google’s London-based sister company DeepMind. Additionally, the tool has predicted nearly complete proteomes for a variety of other organisms, from mice and maize to the malaria parasite.
The accuracy of the more than 350,000 protein structures available through a public database varies. However, researchers believe the resource — which is expected to reach 130 million structures by year’s end — has the potential to revolutionise the life sciences.
From my perspective, it is completely transformative. Having the shapes of all these proteins provides a wealth of information about their mechanisms,” explains Christine Orengo, a computational biologist at University College London (UCL).
“This is the most significant contribution an AI system has made to the advancement of scientific knowledge to date. That is not an exaggeration,” says Demis Hassabis, co-founder and CEO of DeepMind.
However, researchers emphasise that the data dump is only the beginning. They will seek to validate the predictions and, more importantly, to apply them to previously impossible experiments. “It’s an incredible first step to have all this data at that scale,” says David Jones, a computational biologist at University College London who advised DeepMind on an earlier version of AlphaFold.
Proteins are composed of long ribbons of amino acids that self-twist into intricate knots. Understanding how diseases work and developing new drugs—or identifying organisms that can help combat pollution and climate change—requires understanding the shape of a protein’s knot. Determining the shape of a protein takes weeks or months in the laboratory. AlphaFold is capable of predicting shapes down to the atomic level within a day or two.
The new database should make biologists’ lives even easier. While AlphaFold is freely available to researchers, not everyone will want to run the software on their own. “It’s much easier to go to the database and retrieve a structure than it is to run it on your own computer,” says David Baker of the University of Washington’s Institute for Protein Design, whose lab developed RoseTTAFold, a tool for predicting protein structure based on AlphaFold’s approach.
Baker’s team has spent the last few months collaborating with biologists who had previously been unable to determine the shape of proteins they were studying. “There is a lot of pretty cool biological research that has been accelerated significantly,” he explains. A public database containing hundreds of thousands of pre-designed protein shapes should serve as an additional accelerator.
“It appears to be awe-inspiring,” says Tom Ellis, a synthetic biologist at Imperial College London who is excited to try the database. However, he cautions that the majority of the predicted shapes have not been validated in the laboratory.
Predictions that have won awards
Last year, DeepMind stunned the life sciences community when an updated version of AlphaFold swept the biennial CASP protein prediction competition (Critical Assessment of Protein Structure Prediction). Researchers predict the structures of proteins whose structures have been experimentally solved but not yet made public in this long-running competition, which has traditionally been the domain of academics.
Certain predictions made by AlphaFold were consistent with very good experimental models, and some scientists predicted the network’s influence would be epochal. DeepMind released the source code for the latest version of AlphaFold and a detailed description of its development process last week1 (academic teams have already begun using these resources to make useful predictions). DeepMind optimised AlphaFold’s code during the process of preparing it for public release. While some of the CASP predictions took days to compute, they can now be computed in minutes to hours using the updated version of AlphaFold.
With this increased efficiency, the DeepMind team set out to predict the structures of nearly every protein known to exist encoded by the human genome, as well as those of 20 model organisms. The structures are stored in a database at EMBL-EBI (the European Molecular Biology Laboratory European Bioinformatics Institute) in Hinxton, United Kingdom.
Along with the predicted structures, which cover 98.5 percent of known human proteins and a comparable percentage for proteins from other organisms, AlphaFold generated a confidence score for its predictions. “We want to provide a crystal-clear signal to experimentalists and biologists about which parts of the predictions they should trust,” says Kathryn Tunyasuvunakool, a science engineer at DeepMind and the first author of a Nature paper describing the human proteome predictions2. According to Tunyasuvunakool, 58 percent of the human proteome’s predictions for the locations of individual amino acids were accurate enough to be confident in the shape of the protein’s folds. A subset of those predictions — 36% of the total — may be precise enough to detail important atomic features for drug design, such as an enzyme’s active site.
Even less precise predictions may provide insight. According to biologists, a large proportion of human proteins and those of other eukaryotes — organisms with nuclei — contain regions that are inherently disordered and acquire a defined structure only when combined with other molecules. “Many proteins are simply wiggly in solution; they lack a fixed structure,” explains John Jumper, AlphaFold’s lead researcher. Some of the regions predicted by AlphaFold with a low degree of confidence correspond to those that biologists suspect are disordered, according to Pushmeet Kohli, DeepMind’s head of AI for science.
The researchers note that determining how individual proteins interact with other cellular players is one of the most difficult aspects of the AlphaFold predictions. The majority of its predictions for the CASP competition involved independently folding units of a protein called domains. However, the human and other organisms’ proteomes contain proteins with multiple domains that fold semi-independently. Additionally, human cells contain molecules composed of multiple chains of interconnected proteins, such as receptors on cell membranes.
Data deluge
By year’s end, Sameer Velankar, a structural bioinformatician at EMBL-EBI, predicts that the approximately 365,000 structure predictions deposited this week will grow to 130 million — nearly half of all known proteins. The database will be updated as new proteins and improved predictions are identified. “This is not a resource you anticipate having access to,” says Tunyasuvunakool, who is intrigued to see what scientists discover.
AlphaFold and related tools are already being used by researchers to aid in the interpretation of experimental data generated by X-ray crystallography and cryo-electron microscopy. Marcelo Sousa, a biochemist at the University of Colorado Boulder, used AlphaFold to create models of proteins used by bacteria to evade an antibiotic called colistin based on X-ray data. The regions of the experimental model that differed from the AlphaFold prediction were typically regions assigned with low confidence by the software, Sousa observes, indicating that AlphaFold is accurate in predicting its limits.
However, the availability of such a large number of protein structures is likely to result in a “paradigm shift” in biology, according to Mohammed AlQuraishi, a computational biologist at Columbia University in New York City who specialises in protein structure prediction. His field has expended so much time and energy on accurately predicting protein structures on this scale that it has not yet figured out how to best utilise these resources. “Everything that we do today that requires a protein sequence can now be accomplished through protein structure.”
Orengo hopes the database will aid her in her quest to gain a better understanding of the structural constraints on proteins. She has classified a database of known proteins into approximately 5,000’structural families,’ but approximately half of the proteins in the database are excluded because no other protein with a known structure exists. AlphaFold’s predictions, she says, may aid in the discovery of novel shapes. “We’ll get a true sense of how folding space looks.”
Jones anticipates that AlphaFold will spur much soul-searching among biologists about what to do with so many structures — and the ease with which they can be created. “Conferences will be held. Now that we have 130 million models, how does this alter our perspective on biology? It may be that it has no effect,” he says. “I have a feeling it will.”
Reference
DeepMind’s AI predicts structures for a vast trove of proteins. https://doi.org/10.1038/d41586-021-02025-4
Related posts:
![google-gemini-ultra-pro-nano-ai-model]()
A Comprehensive Guide to Google Gemini's Multimodal AI Revolution
A.I![chatgpt]()
Z-DNA Flipons & Disease: How AI is Revolutionizing Genomic Research
news![Decoding the Enigmas of the Y Chromosome: Progress, Significance, and Constraints]()
Decoding the Enigmas of the Y Chromosome: Progress, Significance, and Constraints
bioinformatics![Global Genomics Initiatives]()
Drug Repurposing: A Genomic Approach
A.I![AI-maths-Chatgpt]()
The Future of Artificial Intelligence: A 20-Year Odyssey
A.I![Artificial_Intelligence__AI__Machine_Learning_-_Deeplearning]()
Computational Biology Datasets Suitable For Machine Learning
A.I![Democratization of AI]()
AI Demonstrates High Accuracy in Identifying Rheumatic Heart Disease from Echocardiograms
news![COVID-19-SARS-CoV-2-deletedsequence.]()
Early COVID-19 symptoms vary by age group, according to new research.
news![chatgpt]()
Prompt Engineering in Bioinformatics
A.I![AI and bioinformatics]()
Utilizing ChatGPT in Bioinformatics Education: A Guide to the REFINED Strategy
A.I![A.I Project ideas]()
Top 18 FREE AI Tools for Research and Academic Writing 2024
A.I![AI-studying-tools]()
Top 10 AI Studying Tools to Boost Your Productivity
A.I![biophysics_bioinformatics]()
Focused Ultrasound Proves Promising in Easing Deep-seated Pain
news![AI and bioinformatics research]()
Nvidia and Genentech Collaborate to Accelerate AI-Based Drug Discovery
news![AI-in-education]()
AI in Bioinformatics Market Soars from $2.8 Billion to $49.2 Billion by 2031
A.I![Artificial_Intelligence__AI__Machine_Learning_-_Deeplearning]()
Recent Advances in Deep Learning for Bioinformatics
A.I