Choosing the Right Bioinformatic Tool for Microbiome Analysis
September 10, 2023A Comparative Analysis: The Effect of Bioinformatic Tools on 16S rRNA-Based Microbiome Studies
Introduction:
When it comes to researching microbial communities via 16S rRNA gene sequencing, your choice in computational methods can make a world of difference. A newly published paper took a look at how five key bioinformatic workflows—NG-Tax1, NG-Tax2, QIIME1, QIIME2, and mothur—measure up in generating research outcomes.
What We Discovered:
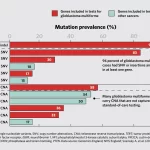
According to the research, NG-Tax1 and NG-Tax2 provided similar outcomes in areas such as the number of reads, singletons, and unidentified reads. When compared to QIIME1, QIIME2, and mothur, the discrepancies were evident. Only 40% of genera were consistent across all platforms, which increased to 70% when applying a 10% prevalence filter. Among these, QIIME2 was most aligned with anticipated microbiome compositions, making it a top choice for this type of study.
A Closer Look at the Tools:
QIIME1:
Even though it’s a bit dated and no longer maintained, this software was still able to identify a wide range of unique microbial taxa.
QIIME2:
This is the go-to tool for those looking for a comprehensive and balanced analysis.
Mothur:
This software generated the highest proportion of singletons, which could muddy the results.
NG-Tax1 & NG-Tax2:
Both of these are particularly stringent when it comes to eliminating artifacts and produced comparable results.
How to Apply These Insights:
The paper offers some clear guidance based on your research focus:
1. If you’re tracking down rare or low-occurrence taxa: Consider **QIIME1 or Mothur**
2. For those of you who want thorough artifact filtering: **NG-Tax1 or NG-Tax2** should be your choice.
3. If you’re after a well-rounded analysis: **QIIME2** is the way to go.
Questions Left to Answer and Study Limits:
The research isn’t perfect, and there are limitations like a modest sample size of 90 and reliance on basic statistical significance, which could introduce inaccuracies. It’s evident that additional studies are needed, especially ones that look at the impact of different datasets and experiment structures.
Wrapping Up:
This recent research emphasizes the importance of selecting the right computational methods for microbiome studies. The tools you choose will influence your study outcomes and how they compare to others in the field. As microbial community studies continue to expand, understanding the strengths and drawbacks of these computational methods becomes all the more critical.
Reference
Szopinska-Tokov J, Bloemendaal M, Boekhorst J, Hermes GD, Ederveen TH, Vlaming P, Buitelaar JK, Franke B, Arias-Vásquez A. A comparison of bioinformatics pipelines for compositional analysis of the human gut microbiome. bioRxiv. 2023:2023-02.
Related posts:
![Superhero of the Cellular World: Talin Protein's Elastic Powers Unveiled]()
Superhero of the Cellular World: Talin Protein's Elastic Powers Unveiled
news![Humangenomeproject-personalizedmedicine]()
Understanding Neurodevelopmental Disorders: Insights from Parental Traits
news![CRISPR-COVID-19]()
CRISPR-based Rapid COVID Diagnostics
news![variantcalling-bioinformatics]()
Researchers Develop New Statistical Tool to Uncover Genetic Causes of Disease
news![CRISPR-COVID-19]()
Duke University Engineers Revolutionize CRISPR Technology for Precision Genome Editing
CRISPR![multiomics]()
The Future of Multi-Omics Integration: A 2043 Vision
bioinformatics![Devin: The World's First Autonomous AI Software Engineer]()
Devin: The World's First Autonomous AI Software Engineer
A.I![protein-structure-analysis-bioinformatics]()
CHRM1 Protein Unveiled as a Therapeutic Target in Advanced Prostate Cancer
news![computationalbiologyandmedicine]()
Scientists Unveil Intricate Neural Landscape of the Hippocampus
news![Genome analysis tools]()
Ancestral Genome Reconstruction Illuminates the World of Degenerate Transposable Elements
news![Stargate-AI]()
The Stargate Project: A $500 Billion Leap into the Future of AI and Its Transformative Impact on Hea...
A.I![AI and bioinformatics research]()
Nvidia and Genentech Collaborate to Accelerate AI-Based Drug Discovery
news![protein-molecular-dynamics]()
Revolutionizing Protein Function Prediction: The Introduction of DeepGO-SE
news![The Largest Whole-genome Sequencing Study in Cancer Unveils Potential Treatment and Prognostic Insig...]()
The Largest Whole-genome Sequencing Study in Cancer Unveils Potential Treatment and Prognostic Insig...
news![cloudstorage-bioinformatics]()
The Future of Cloud Computing and Big Data Management: A 2043 Horizon
bioinformatics![Researchers unlock the chronic kidney disease 'treasure map']()
Researchers unlock the chronic kidney disease 'treasure map'
news