

Modelling a protein through automatic comparative modelling servers
May 11, 2007>> Choosing a suitable template sequences
>> Algining template and target sequences
>> Building backbone
>> constructing loop and generating side chains
Finding suitable template is usually done by performing a BLAST search against the sequences in the PDB, the respoistory for protein sturcture obtained by X-ray crystallography and NMR. All sequences in the PDb with an E-value of Blast below a certain treshold are considered as candidates for the template. The alignment between template and target sequences should atleast contain 30% identities and most important it should have existence of conserved regions. A mulitple sequence alignment with same family members of the template or several template sequences may also be constructed. This step requires some manual correction of the alignment in order to obtain a reliable model.Several methods are used for loop building and side chain reconstruction.
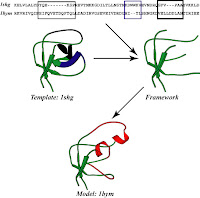
When no suitable template is available for comparative modeling, de novo modeling methods (also called ab initio modeling) may be used. The success rate with such modeling is considerably lower than that with comparative modeling. The accuracy of de novo models is too low for problems requiring high-resolution structure information.”
Automatic Comparative modelling servers
Related posts:
![Orf]()
Difference Between CDS and ORF: A Beginner’s Guide to Bioinformatics
bioinformatics![Artificial Intelligence in Pharma]()
Why do pharmaceutical companies employ bioinformaticians?
bioinformatics![bioinformatics-Space]()
Beyond Earth: Bioinformatics, the Unsung Hero of Space Exploration
bioinformatics![machinelearning_omicstutorials]()
Bioinformatics and Machine Learning: From Sequence Analysis to Predictive Modeling
A.I![python-bioinformatics]()
Python via Bioinformatics Examples
bioinformatics![protein-structure-analysis-bioinformatics]()
Recent Advances in Structural Biology and Structural Bioinformatics
bioinformatics![Career Paths-Future]()
Navigating the Future of Bioinformatics: Trends, Innovations, and Key Players
bioinformatics![machinelearning-omicsfield]()
Introduction to Machine learning-Bioinformatics
bioinformatics![systembiology]()
Systems Biology and Network Analysis: Interactions and Pathways
bioinformatics![and Transcriptomics]()
An Introduction to Proteomics, Metabolomics, and Transcriptomics
bioinformatics![bioinformatics jobs]()
Step-by-Step Manual: Will a Master’s in Bioinformatics Help in Getting a Job?
bioinformatics![remotecomputer-bioinformatics]()
Step-by-Step Guide: Creating Venn/Euler Diagrams for Six or More Sets in Bioinformatics
bioinformatics![proteomics-omics]()
A Comprehensive Guide to Python Programming for PDB Analysis in Bioinformatics
bioinformatics![Bioinformatics]()
Unlocking the Future: How Genome Sequencing is Transforming Bioinformatics and Revolutionizing Medic...
bioinformatics![blastquery]()
Basic Local Alignment Search Tool (BLAST) for bioinformatics
bioinformatics![python-bioinformatics-basics]()
Step-by-Step Guide: Removing Duplicate Sequences in FASTA Files
bioinformatics















