

Motif- Sequence & Structure motifs-Bioinformatics analysis
July 22, 2019Motif
A short (usually not more than 20 amino acids) conserved sequence of biological significance.
Motif are of two types (1) Sequence motifs and (2) structure motifs
Motif discovery is the problem of finding recurring patterns in biological data. Patterns can be sequential, mainly when discovered in DNA sequences. They can also be structural (e.g. when discovering RNA motifs). Finding common structural patterns helps to gain a better understanding of the mechanism of action (e.g. post-transcriptional regulation). Unlike DNA motifs, which are sequentially conserved, RNA motifs exhibit conservation in structure, which may be common even if the sequences are different.
Sequence motifs
A sequence motif is a nucleotide or amino-acid sequence pattern that is widespread and has, or is conjectured to have, a biological significance. A protein sequence motif is an amino-acid sequence pattern found in similar proteins; change of a motif changes the corresponding biological function.
Motifs occurrence
When a sequence motif appears in the exon of a gene, it may encode the “structural motif” of a protein; that is a stereotypical element of the overall structure of the protein. Nevertheless, motifs need not be associated with a distinctive secondary structure. “Noncoding” sequences are not translated into proteins, and nucleic acids with such motifs need not deviate from the typical shape (e.g. the “B-form” DNA double helix).
Outside of gene exons, there exist regulatory sequence motifs and motifs within the “junk”, such as satellite DNA. Some of these are believed to affect the shape of nucleic acids (see for example RNA self-splicing), but this is only sometimes the case. For example, many DNA binding proteins that have affinity for specific DNA binding sites bind DNA in only its double-helical form. They are able to recognize motifs through contact with the double helix’s major or minor groove

Short coding motifs, which appear to lack secondary structure, include those that label proteins for delivery to particular parts of a cell, or mark them for phosphorylation.
Specific sequence motifs usually mediate a common function, such as protein-binding or targeting to a particular subcellular location, in a variety of proteins.
Due to their short length and high level of sequence variability most motifs cannot be reliably predicted by computational means. Therefore we only annotate putative motifs when there is experimental evidence that the motif is functionally important or the presence of the putative motif is consistent with the function of the protein.

Structural motifs
Structural motifs are short segments of protein 3D structure, which are spatially close but not necessarily adjacent in the sequence. Structural motifs may be conserved in a large number of different proteins. Their role may be structural or functional.

Structural motif examples
1.Zinc finger motif
Zinc fingers are a common motif in DNA-binding proteins. The fingers usually are organized as a single series of tandem repeats; occasionally there is more than one group of fingers
2.Helix-turn-helix
Helix-turn-helix motif which can bind DNA. This is a structural feature that is difficult to identify from the amino acid sequence alone
3.Four-helix bundle motif
This structural motif has been observed both as an isolated three-dimensional fold and as a domain within much larger and more complex protein structures. The helices of four-helix bundles are longer than average (~20 residues, as opposed to the usual 10) and are amphipathic, with hydrophobic residues buried in the core. The antiparallel packing of the helices in the bundle may be favored by interaction between the helix dipoles.
4.Greek motif
The Greek key motif consists of four adjacent antiparallel strands and their linking loops. It consists of three antiparallel strands connected by hairpins, while the fourth is adjacent to the first and linked to the third by a longer loop. This type of structure forms easily during the protein folding process.It was named after a pattern common to Greek ornamental artwork.
5.Beta-turn
A beta turn consists of four consecutive residues where the polypeptide chain folds back on itself by nearly 180 degrees
Common motifs
There are certain motifs that occur over and over again in different proteins. The helix-loop-helix motif, for example, consists of two α helices joined by a reverse turn. The Greek key motif consists of four antiparallel β strands in a β sheet where the order of the strands along the polypeptide chain is 4, 1, 2, 3. The β sandwich is two layers of β sheet.
The vast majority of motifs do not have a common evolutionary origin in spite of many claims to the contrary. They arise independently and converge on a common stable structure. The fact that these same motifs occur in hundreds of different proteins indicates that there are a limited number of possible folds in the universe of protein structures. The original primitive protein may have been relatively unstructured but over time there will be selection for more and more stable structures.

Larger motifs
Larger motifs are often called domain folds because they make up the core of a domain. The parallel twisted sheet is found in many domains that have no obvious relationship other than the fact that they share this very stable core structure. The β barrel structure is found in many membrane proteins. There are dozens of enzymes that have adapted to an α/β barrel. These enzymes are not evolutionarily related. (The β helix is much less common.)

The term motif is used in two different ways in structural biology. The first refers to a particular amino-acid sequence that is characteristic of a specific biochemical
function.
example: CXX(XX)CXXXXXXXXXXXXHXXXH
Sequence motifs can be recognized by inspecting the amino-acid sequence. Databases of such motifs exist:
• e.g., PROSITE (http://www.expasy.ch/prosite/)
The second use of the term motif refers to a set of contiguous secondary structure elements that have a particular functional significance.
e.g. helix-turn-helix, Greek-key motif
Usually, sequence motifs are more indicative of certain
function, because a shared structural motif does not always imply similar function. However, detecting functional motifs from sequence alone is difficult due to variable spacing, different ordering of functional residues.
Pattern
Qualitative expression of a motif
Regular Expression – C[TA]TTG{X}
Profile
Quantitative expression of a motif
Position Specific Scoring Matrices (PSSMs), Weight matrices
What is the difference between motifs and domains?
A motif is similar 3-D structure conserved among different proteins that serves a similar function. An example is a helix-turn-helix motif. This is a structure that is seen in unrelated proteins that bind DNA, so the presence of a helix-turn-helix motif is an indication of a protein’s function.
Domains, on the other hand, are regions of a protein that has a specific function and can (usually) function independently of the rest of the protein. A protein which has multiple domains. It has a DNA binding domain located towards the N terminus of the protein, and a catalytic domain that is located closer to the C-terminus. Theoretically you can separate the domains from each other and the DNA binding domain will still bind DNA and the catalytic domain will still perform catalysis. There is some overlap with the definitions of domain and motif. Some motifs are also considered domains, and vice versa.
Interpreting the Biological Role of Motifs
Once an interesting set of motifs has been identified by motif discovery, the next logical step is to interpret the biological role of these sequence features. It may be possible to associate motifs with specific observable effects like up-regulation or down-regulation of gene expression in certain experimental conditions. Further biological insight into regulatory networks can be obtained by associating specific transcription factors with the motifs to which they bind. Standard motif discovery tools do not directly address these issues of interpretation. However, more recently, techniques have been developed that explore these questions.
Bioinformatics tools and databases for motif identification:
Many excellent resources are available for analyzing sequence data with motif discovery, postprocessing motifs, and obtaining sequence and motif data. Freely available packages exist that integrate multiple motif discovery tools, and can greatly facilitate motif discovery analyses. Many stand-alone motif discovery tools are available in downloadable and Web-enabled form. Tools for motif scanning are often available with prepackaged libraries of known motifs, but also allow scans with custom motifs learned by motif discovery

Sequence motif algorithms
Common algorithms used in motif identification are
• Enumeration
• Probabilistic Optimization
• Deterministic Optimization
Enumeration
• Exhaustive search: word counting method, count all n-mers and look for overrepresentation
• Less likely to get stuck in a local optimum
• Computationally expensive
YMF
http://wingless.cs.washington.edu/YMF/YMFWeb/YMFInput.pl
Weeder
http://159.149.160.88/modtools/
Probabilistic Optimization
• Uses a Gibbs sampling approach
• One n-mer from each sequence is randomly picked to determine initial model. In subsequent iterations, one sequence, i, is removed and the model is recalculated. Pick a new location of motif in sequence i iterate until convergence
• Assumes most sequences will have the motif
AlignAce
http://www1.spms.ntu.edu.sg/~chenxin/W-AlignACE/
Gibbs Motif Sampler
http://bayesweb.wadsworth.org/gibbs/gibbs.html
Deterministic Optimization
• Based on expectation maximization (EM)
• EM: iteratively estimates the likelihood given
the data that is present
I. Expectation step: Use current parameters (and
observations) to reconstruct hidden structure
II. Maximization step: Use that hidden structure (and
observations) to re-estimate parameters
Structure motifs algorithms
FOLDALIGN-
Based on Sankoff’s algorithm. It maximizes alignment similarity and number of base pairs formed in 2 aligned sequences.
FOLDALIGN- http://foldalign.ku.dk/software/index.html
SLASH
Uses FOLDALIGN to find local alignments in RNA sequences. Then COVE , to build a SCFG model from the local alignments.
Mauri & Pavesi
Uses Affix trees for the discovery of hairpins, bulges and internal loops in RNA. Substrings of certain length appearing in at least q sequences are found and expanded.
CMfinder
Based on expectation maximization (EM) to simultaneously align and fold sequences using covariance model of RNA motifs.
CMfinder – http://bio.cs.washington.edu/yzizhen/CMfinder/
RNAProfile
Uses a heuristic to extract a set of candidate regions from each sequence. The second step involves grouping regions to find similar motifs.
RNAProfile- http://159.149.160.51/modtools/downloads/rnaprofile.html
RNAPromo
The motif prediction algorithm initially looks for structural elements which are common to the input RNAs, and then employs an expectation maximization algorithm to refine the resulting probabilistic model.
RNAPromo- http://genie.weizmann.ac.il/pubs/rnamotifs08/index.html
Case Study:
Sequence motif prediction
CDART: Conserved Domain Architecture Retrieval Tool. This program gives an interactive graphical display of the conserved motifs found in an amino acid sequence. You can click on each domain to learn more about its properties and consensus sequence. The program also provides graphical displays of all known proteins containing at least one of the domains found in your protein. One drawback is that this program only reports major domains, and not smaller motifs, and has fairly brief descriptions. 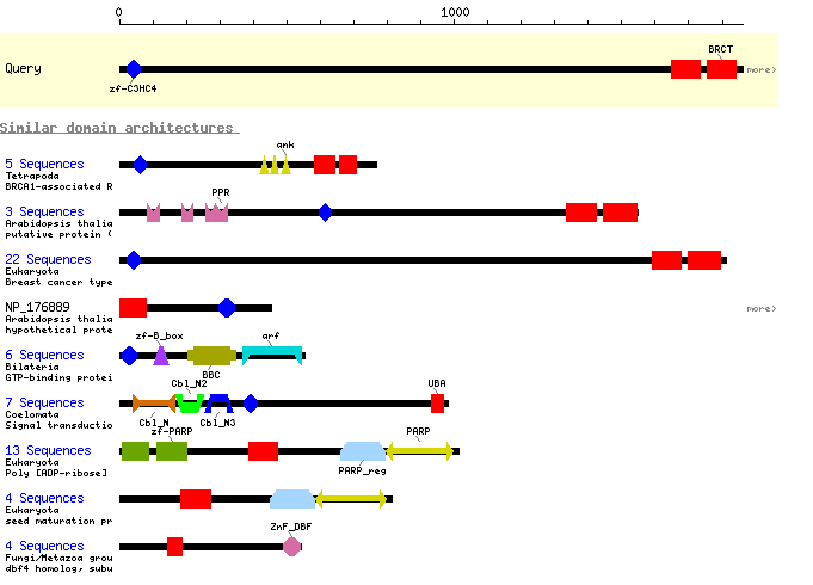
1. The program PROSITE analyzes a protein sequence for these known motifs and gives a description of each. This is useful when analyzing the sequence of a new protein to try to gain clues to its function.
https://prosite.expasy.org/scanprosite/
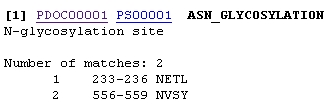
Enter the amino acid sequence that you wish to analyze or the accession number of the protein and press Start the Scan. You will be given an output which lists several motifs present in the protein, indicating the sequence that was identified and its position in the protein. Each will also contain a link to more information on that particular motif.
For example the sequence being analyzed has potential N-glycosylation sites at amino acids 233 and 556. By clicking on PDOC00001 more information on N-glycosylation will be provided.
 |
Other motifs are more complex and can include sites that bind cofactors or substrates (active site). Such information would be valuable in identifying the function of a protein.
 |
References:
1.MacIsaac, K. D., & Fraenkel, E. (2006). Practical strategies for discovering regulatory DNA sequence motifs. PLoS computational biology, 2(4), e36.
2.Badr, G., Al-Turaiki, I., & Mathkour, H. (2013). Classification and assessment tools for structural motif discovery algorithms. BMC bioinformatics, 14(9), S4.
Related posts:
![3Dstructureofprotein-deepmind]()
Frequently asked questions (FAQ's) about Alpha Fold
bioinformatics![translation bioinformatics]()
Alternative Splicing in Disease: Bioinformatics Approaches
bioinformatics![protein-structure-analysis-bioinformatics]()
How to Avoid Conversion of Gene Symbols to Date Format in Excel
bioinformatics![Spatial Metabolomics]()
Challenges in Metabolomics Bioinformatics
bioinformatics![A-RNA-sequence-analysis-basics.]()
Step-by-Step Guide: Make your first bioinformatics project
bioinformatics![bioinformatics-trend-2024]()
Biomedical Informatics vs Bioinformatics: Understanding the Difference
bioinformatics![Bioinformatics glossary - U]()
Bioinformatics glossary - U
bioinformatics![bioinformatics-fileformat-basics]()
Best Practices for Handling Very Large Datasets in Bioinformatics
bioinformatics![bioinformatics-FAQ]()
Frequently asked questions about bioinformatics
bioinformatics![bioinformatics software]()
Blockchain Technology in Genomics for Enhanced Data Ownership and Integrity
bioinformatics![AI-drug discovery]()
How Artificial Intelligence is Revolutionizing Drug Discovery and Healthcare
A.I![Quantum_Computing_bioinformatics-omicstutorials]()
Introduction to Quantum Computing
bioinformatics![Earthbiogenomeproject_omicstutorials]()
Earth BioGenome Project (EBP) – Exploring the Dark Matter of Biology
bioinformatics![blastquery]()
Basic Local Alignment Search Tool (BLAST) for bioinformatics
bioinformatics![cancer-bioinformatics]()
Bioinformatics Tools for Precision Oncology: A Comprehensive Guide to Genomic Biomarker Discovery
bioinformatics![bioinformatics jobs]()
Climbing the Career Ladder in Bioinformatics: Skills and Tips for Getting Ahead
bioinformatics

















