Unlock Deeper Proteomics Insights with Advanced Mass Spectrometry
November 29, 2023I. Introduction to Mass Spectrometry for Proteomics
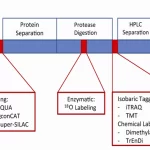
A. Overview of MS-Based Proteomics Workflow
1. Introduction to Proteomics:
- Definition:
- Proteomics is the large-scale study of proteins in a biological system, involving the identification, quantification, and characterization of proteins.
- Significance:
- Provides insights into cellular processes, functions, and interactions at the protein level.
2. Role of Mass Spectrometry (MS) in Proteomics:
- Definition of Mass Spectrometry:
- Mass spectrometry is an analytical technique used to measure the mass-to-charge ratio of ions, enabling the identification and quantification of biomolecules.
- Application in Proteomics:
- In proteomics, MS is a central tool for analyzing proteins by measuring the mass of peptides generated through enzymatic digestion.
3. Key Components of MS-Based Proteomics Workflow:
a. Sample Preparation:
- Process involving protein extraction, digestion into peptides, and purification to prepare samples for MS analysis.
b. Liquid Chromatography (LC):
- Separation of peptides based on properties such as size, charge, and hydrophobicity before entering the MS instrument.
c. Mass Spectrometer:
- Instrument for ionizing peptides, measuring their mass-to-charge ratio, and generating mass spectra for further analysis.
d. Data Analysis:
- Interpretation of mass spectra to identify peptides and proteins, involving database searching and bioinformatics tools.
4. Protein Identification and Quantification:
a. Database Searching:
- Matching experimental mass spectra to theoretical spectra generated from protein databases to identify proteins.
b. Label-Free and Isotope Labeling Quantification:
- Methods for quantifying protein abundance based on MS signal intensities, either with or without the use of stable isotopes.
5. Challenges and Advances in MS-Based Proteomics:
a. Sensitivity and Dynamic Range:
- Challenges in detecting low-abundance proteins and expanding the dynamic range of detection.
b. Advances in Instrumentation:
- Ongoing developments in MS instrumentation, such as high-resolution MS and tandem MS, to address sensitivity and dynamic range challenges.
c. Integration with Multi-Omics Approaches:
- Increasing integration of proteomics with genomics, transcriptomics, and metabolomics for a comprehensive understanding of biological systems.
6. Applications of MS-Based Proteomics:
a. Biomarker Discovery:
- Identification of protein biomarkers for disease diagnosis, prognosis, and therapeutic response.
b. Drug Discovery:
- Assessment of drug targets, mechanisms of action, and evaluation of drug efficacy.
c. Functional Proteomics:
- Understanding protein function, post-translational modifications, and cellular signaling pathways.
7. Future Directions in MS-Based Proteomics:
a. Single-Cell Proteomics:
- Advancements towards analyzing proteomes at the single-cell level for enhanced resolution.
b. Quantitative Phosphoproteomics:
- Improvements in quantifying phosphorylation events to understand dynamic cellular signaling.
c. Integration with Structural Proteomics:
- Combining MS-based proteomics with structural techniques for a more comprehensive characterization of proteins.
The MS-based proteomics workflow is a powerful and versatile approach for unraveling the complexity of cellular proteomes. As technology advances, MS continues to play a pivotal role in driving discoveries in various fields, from basic research to clinical applications.
II. Maximizing Protein Identification Through Tandem MS
A. Strategies for Optimal Peptide Fractionation
1. Introduction to Peptide Fractionation:
- Definition:
- Peptide fractionation involves separating complex peptide mixtures into simpler fractions before MS analysis, improving coverage and depth of proteome analysis.
2. Common Strategies for Peptide Fractionation:
a. Liquid Chromatography (LC):
- Use of high-performance liquid chromatography to separate peptides based on physicochemical properties such as hydrophobicity.
b. Strong Cation Exchange (SCX):
- Fractionation based on peptide charge, where peptides are separated using a column with varying ionic strengths.
c. Electrophoresis Techniques:
- Gel-based (e.g., SDS-PAGE) or gel-free techniques for size-based separation of peptides.
d. High-pH Reversed-Phase Fractionation:
- Separation of peptides at high pH, providing an alternative to low-pH reversed-phase LC.
3. Benefits of Peptide Fractionation:
- Increased Dynamic Range:
- Enables the identification of low-abundance peptides by reducing the complexity of the sample.
- Enhanced Sensitivity:
- Improves sensitivity by focusing on specific peptide subsets, increasing the likelihood of detecting less abundant proteins.
B. Novel Dissociation Techniques like EThcD
1. Introduction to EThcD:
- Definition:
- Electron-Transfer/Higher-Energy Collision Dissociation (EThcD) is a hybrid fragmentation technique that combines electron transfer dissociation (ETD) and higher-energy collision dissociation (HCD) to enhance peptide sequencing.
2. Advantages of EThcD:
a. Complementary Fragmentation:
- Provides complementary fragmentation information, improving the identification of peptides and post-translational modifications.
b. Improved Sequence Coverage:
- Enhances sequence coverage, particularly for larger peptides and proteins.
c. Selective Activation:
- Allows selective activation of specific peptide ions, leading to a more targeted and informative dissociation.
C. Interpreting Tandem MS Data
1. Database Searching for Peptide Identification:
- Algorithmic Approaches:
- Use of database search algorithms (e.g., SEQUEST, Mascot) to match experimental tandem MS spectra with theoretical spectra generated from protein databases.
2. Key Considerations in Interpreting Tandem MS Data:
a. Peptide Sequence Matching:
- Matching observed fragment ions with theoretical ions to assign peptide sequences.
b. Post-Translational Modification Identification:
- Identification of post-translational modifications by analyzing characteristic mass shifts in tandem MS spectra.
c. False Discovery Rate (FDR) Control:
- Implementation of FDR control to minimize the chances of false-positive identifications.
d. Spectral Quality Assessment:
- Assessment of the quality of tandem MS spectra, considering factors such as signal-to-noise ratio and peak intensity.
3. Utilizing Bioinformatics Tools:
a. Peptide Spectrum Matching (PSM) Algorithms:
- Implementation of PSM algorithms for accurate assignment of peptides to tandem MS spectra.
b. Protein Inference:
- Strategies for protein inference to identify the most likely proteins associated with the observed peptides.
c. Quantitative Proteomics:
- Integration of quantitative information from tandem MS data for protein abundance analysis.
The strategic use of peptide fractionation, innovative dissociation techniques like EThcD, and the informed interpretation of tandem MS data are essential for maximizing protein identification in MS-based proteomics. These advancements contribute to a more comprehensive understanding of the proteome, facilitating discoveries in biology, medicine, and drug development.
III. Quantitative Proteomic Profiling Approaches
A. Label-Free Quantification (LFQ)
1. Overview of Label-Free Quantification:
- Definition:
- Label-Free Quantification (LFQ) is a method for assessing protein abundance without the use of chemical labels. It relies on the direct comparison of peak intensities or spectral counts between different samples.
2. Principles of LFQ:
a. Peak Intensity Comparison:
- Comparison of the intensity of peptide peaks in MS spectra between different samples.
b. Spectral Counting:
- Counting the number of MS/MS spectra identifying a particular peptide as a measure of its abundance.
3. Advantages of LFQ:
a. Simplicity and Cost-Effectiveness:
- LFQ does not require additional labeling steps, making it simpler and more cost-effective.
b. Dynamic Range:
- Suitable for a broad dynamic range of protein abundances.
c. Flexibility:
- Applicable to various sample types and experimental setups.
B. Isotope Labeling Methods like TMT/iTRAQ
1. Introduction to Isotope Labeling:
- Definition:
- Isotope labeling methods, such as Tandem Mass Tag (TMT) and Isobaric Tags for Relative and Absolute Quantification (iTRAQ), involve chemically labeling peptides from different samples with isotope-coded tags.
2. Principles of Isotope Labeling:
a. Multiplexing:
- TMT and iTRAQ enable the simultaneous analysis of multiple samples within a single mass spectrometry experiment, allowing for increased throughput.
b. Quantification via Reporter Ions:
- Isotope-labeled tags generate reporter ions during MS/MS fragmentation, and the intensity of these ions is used for quantification.
3. Advantages of Isotope Labeling:
a. Precise Quantification:
- Provides precise and accurate quantification due to the isobaric nature of the tags.
b. Multiplexing Capability:
- Enables the comparison of multiple samples in a single experiment, reducing technical variability.
c. Consistent Workflow:
- Standardized labeling protocols contribute to consistent and reproducible workflows.
C. Absolute Quantification Strategies
1. Introduction to Absolute Quantification:
- Definition:
- Absolute quantification involves determining the absolute abundance of specific proteins or peptides in a sample by comparing them to known standards.
2. Principles of Absolute Quantification:
a. Stable Isotope-Labeled Peptide Standards:
- Incorporation of stable isotope-labeled peptides as internal standards for accurate quantification.
b. Selected Reaction Monitoring (SRM) or Parallel Reaction Monitoring (PRM):
- Targeted mass spectrometry approaches for precise quantification of specific peptides.
3. Advantages of Absolute Quantification:
a. Accuracy and Precision:
- Provides high accuracy and precision in quantifying targeted proteins or peptides.
b. Comparison Across Experiments:
- Facilitates comparison of protein abundance across different experiments and conditions.
c. Normalization of Data:
- Allows for normalization of data, minimizing variations introduced during sample preparation and analysis.
4. Applications:
- Clinical Biomarker Validation:
- Useful for validating potential clinical biomarkers by precisely quantifying their abundance.
- Pharmacokinetic Studies:
- Applied in pharmacokinetic studies to determine drug concentrations in biological samples.
- Targeted Protein Quantification:
- Suitable for targeted quantification of specific proteins with known biological relevance.
Quantitative proteomic profiling approaches, including Label-Free Quantification (LFQ), isotope labeling methods like TMT/iTRAQ, and absolute quantification strategies, offer diverse options for accurately assessing protein abundance. The choice of method depends on experimental goals, sample complexity, and the level of precision required for the study.
IV. Achieving In-Depth Proteome Coverage
A. Deep, Unbiased Analysis with Ultra-High Resolution Mass Spectrometry
1. Introduction to Ultra-High Resolution Mass Spectrometry:
- Definition:
- Ultra-high resolution mass spectrometry (UHR-MS) involves using instruments with exceptionally high resolving power, allowing for the precise measurement of mass-to-charge ratios of ions.
2. Advantages of UHR-MS for Proteome Analysis:
a. Enhanced Spectral Resolution:
- Higher resolution enables the separation of closely spaced peaks, reducing spectral overlap and improving accuracy in mass measurements.
b. Increased Dynamic Range:
- UHR-MS extends the dynamic range, facilitating the detection of both high and low-abundance proteins.
c. Improved Peptide Identification:
- Better discrimination of isobaric species enhances the accuracy of peptide and protein identifications.
3. Applications in Achieving In-Depth Proteome Coverage:
a. Characterization of Complex Proteomes:
- Particularly beneficial for analyzing complex proteomes where a high level of detail and precision is required.
b. Quantitative Proteomics:
- Contributes to accurate quantification by reducing interference from co-eluting peptides.
c. Post-Translational Modification Analysis:
- Facilitates the identification and quantification of post-translational modifications with increased confidence.
B. Optimized Protein Extraction Protocols
1. Importance of Protein Extraction:
- Rationale:
- Efficient protein extraction is crucial for obtaining a representative sample that reflects the entire proteome of interest.
2. Key Considerations for Protein Extraction Protocols:
a. Compatibility with Downstream Analysis:
- Protocols should be tailored to the specific requirements of subsequent MS analysis, ensuring compatibility with different MS workflows.
b. Inclusion of Detergents and Solubilization Agents:
- Use of detergents and solubilization agents to efficiently extract membrane proteins and hydrophobic peptides.
c. Minimization of Sample Contamination:
- Strategies to minimize contamination from non-proteinaceous substances, ensuring purity in the extracted protein sample.
3. Innovations in Protein Extraction:
a. Single-Cell Proteomics:
- Protocols optimized for single-cell proteomics to extract proteins from individual cells for detailed analysis.
b. Subcellular Fractionation:
- Fractionation techniques to isolate proteins from specific cellular compartments, enabling targeted analysis.
C. Subcellular Analysis Specificity
1. Subcellular Fractionation Techniques:
- Definition:
- Subcellular fractionation involves separating cellular components into distinct fractions based on their properties, such as size, density, or affinity.
2. Applications of Subcellular Analysis:
a. Enrichment of Subcellular Proteomes:
- Enables the enrichment of proteins from specific cellular compartments, providing insights into localized protein functions.
b. Identification of Organelle-Specific Proteins:
- Facilitates the identification of proteins localized to specific organelles or cellular structures.
c. Improved Detection of Low-Abundance Proteins:
- Reduces sample complexity, enhancing the detection of low-abundance proteins that may be masked in whole-cell lysates.
3. Technological Advances in Subcellular Analysis:
a. Quantitative Subcellular Proteomics:
- Integration of subcellular fractionation with quantitative proteomics for accurate profiling of organelle-specific proteomes.
b. Live-Cell Imaging Coupled with Proteomics:
- Combining live-cell imaging techniques with proteomics for real-time visualization and analysis of subcellular dynamics.
Achieving in-depth proteome coverage involves leveraging advanced technologies such as ultra-high resolution mass spectrometry, optimizing protein extraction protocols, and implementing subcellular analysis specificity. These strategies enhance the depth and precision of proteomic analyses, providing a comprehensive view of the cellular proteome and facilitating discoveries in diverse biological contexts.
V. Emerging High-Throughput Workflows
A. Novel Separation Polymers and NanoPotion
1. Introduction to Novel Separation Polymers:
- Definition:
- Novel separation polymers refer to advanced materials used in chromatographic techniques to enhance the separation of peptides and proteins.
2. Advancements in Separation Polymers:
a. Improved Chromatographic Resolution:
- Utilization of novel polymers with enhanced selectivity and efficiency for improved chromatographic separation.
b. Compatibility with Mass Spectrometry:
- Compatibility with MS-friendly conditions to ensure optimal ionization and detection during mass spectrometry analysis.
3. NanoPotion Technology:
a. Definition of NanoPotion:
- NanoPotion technology involves the use of nanomaterials, such as nanoparticles, to enhance sample preparation and separation efficiency in proteomic workflows.
b. Applications in High-Throughput Workflows:
- Facilitates rapid and efficient sample preparation for high-throughput proteomic analyses.
c. Minimization of Sample Loss:
- NanoPotion technology aims to minimize sample loss, allowing for the analysis of low-abundance proteins.
B. AI for Spectral Analysis Automation
1. Integration of Artificial Intelligence (AI) in Proteomics:
- Rationale:
- AI is being increasingly employed to automate and streamline various aspects of proteomic data analysis.
2. Applications of AI in Spectral Analysis:
a. Peptide Identification:
- AI algorithms aid in accurate and rapid peptide identification by analyzing mass spectra and matching them to peptide sequences.
b. Quality Control:
- Automated AI-based tools for quality control of mass spectrometry data, ensuring the reliability of results.
c. Data Interpretation:
- AI-driven tools assist in the interpretation of complex mass spectra, providing insights into protein modifications and structural features.
3. Benefits of AI in High-Throughput Proteomics:
a. Increased Speed and Efficiency:
- Accelerates data analysis processes, reducing the time required for high-throughput experiments.
b. Enhanced Accuracy:
- Improves the accuracy of spectral interpretation and protein identification, reducing false positives and negatives.
c. Adaptability to Diverse Experimental Conditions:
- AI models can adapt to diverse experimental conditions, making them versatile for different high-throughput workflows.
C. Multiplexing Advances with TMTpro
1. Introduction to TMTpro:
- Definition:
- Tandem Mass Tag (TMT) technology has evolved with the introduction of TMTpro, a multiplexing reagent for quantitative proteomic analysis.
2. Advancements in Multiplexing with TMTpro:
a. Increased Multiplexing Capacity:
- TMTpro offers expanded multiplexing capabilities, allowing for the simultaneous analysis of a larger number of samples in a single experiment.
b. Enhanced Isobaric Tag Design:
- Improved isobaric tag design for more accurate and reliable quantification of proteins across multiple samples.
c. Quantitative Accuracy:
- TMTpro enhances quantitative accuracy by minimizing reporter ion interference and improving dynamic range.
3. Applications in High-Throughput Proteomics:
a. Large-Scale Comparative Studies:
- Enables large-scale comparative studies by multiplexing samples, reducing experimental variability.
b. Time and Resource Efficiency:
- Enhances efficiency in terms of time and resources by analyzing multiple samples simultaneously.
c. Quantitative Analysis in Diverse Biological Contexts:
- Applicable to quantitative analysis in various biological contexts, including clinical, environmental, and systems biology studies.
Emerging high-throughput workflows in proteomics leverage innovations such as novel separation polymers and NanoPotion for efficient sample preparation, the integration of AI for automated spectral analysis, and multiplexing advances with TMTpro to enable simultaneous analysis of multiple samples. These technologies contribute to the scalability, speed, and accuracy of high-throughput proteomic experiments, opening new avenues for comprehensive biological insights.
VI. Clinical and Translational Proteomics Frontiers
A. Cancer Proteogenomics Innovations
1. Integration of Proteomics and Genomics in Cancer Research:
- Rationale:
- Cancer proteogenomics involves the integration of proteomic and genomic data to gain a holistic understanding of cancer biology and identify potential therapeutic targets.
2. Key Innovations in Cancer Proteogenomics:
a. Comprehensive Molecular Characterization:
- Simultaneous analysis of genomic alterations and proteomic changes to achieve a more comprehensive molecular characterization of tumors.
b. Identification of Neoantigens:
- Discovery of tumor-specific neoantigens through proteogenomic analysis, informing the development of personalized cancer immunotherapies.
c. Phosphoproteomics for Signaling Pathways:
- Application of phosphoproteomics to decipher aberrant signaling pathways in cancer cells, aiding in the identification of targeted therapies.
3. Clinical Implications:
a. Patient Stratification:
- Integration of proteogenomic data for patient stratification, allowing for more precise treatment decisions based on individual tumor profiles.
b. Predictive Biomarkers:
- Identification of predictive biomarkers for treatment response and resistance through a comprehensive analysis of protein and genomic alterations.
c. Therapeutic Target Discovery:
- Uncovering novel therapeutic targets by understanding the interplay between genomic mutations and proteomic changes in cancer cells.
B. Autoantibody Profiling for Biomarkers
1. Introduction to Autoantibody Profiling:
- Definition:
- Autoantibody profiling involves the systematic analysis of autoantibodies present in the blood or serum, offering insights into autoimmune responses and disease-associated antigens.
2. Advancements in Autoantibody Profiling:
a. High-Throughput Screening Platforms:
- Utilization of high-throughput screening platforms, such as protein microarrays, for the simultaneous detection of a large number of autoantibodies.
b. Multiplexed Assays:
- Development of multiplexed assays to profile multiple autoantibodies concurrently, enhancing the efficiency of biomarker discovery.
c. Integration with Proteomic Data:
- Integration of autoantibody profiling with proteomic data to identify specific antigens associated with autoimmune responses in various diseases.
3. Clinical Applications:
a. Early Detection of Diseases:
- Autoantibody profiling as a tool for early detection of diseases, including autoimmune disorders and certain cancers, improving prognosis and treatment outcomes.
b. Biomarker Discovery and Validation:
- Identification and validation of autoantibody biomarkers for different diseases, contributing to the development of diagnostic assays.
c. Monitoring Disease Progression:
- Monitoring changes in autoantibody profiles to track disease progression and assess the effectiveness of therapeutic interventions.
4. Translational Impact:
a. Personalized Medicine Approaches:
- Implementation of personalized medicine approaches based on the unique autoantibody profiles of individuals.
b. Precision Diagnostics:
- Contribution to precision diagnostics by integrating autoantibody data with other omics information, such as genomics and proteomics.
The frontiers of clinical and translational proteomics encompass innovations in cancer proteogenomics, where the integration of proteomic and genomic data enhances our understanding of cancer biology and informs personalized treatment strategies. Additionally, autoantibody profiling emerges as a powerful tool for biomarker discovery and personalized diagnostics across various diseases, providing valuable insights into autoimmune responses and disease-associated antigens.
Related posts:
![Proteomics-Quick-Study-A-Brief-Introduction]()
Proteomics Quick Study: A Brief Introduction
Guides![Neuroproteomics]()
Proteomics: Methods & Applications
Guides![Proteomics tools]()
How are proteomics datasets analyzed to reveal protein regulation?
proteomics![AlphaFold 3 and Protein Structure Prediction: Transforming Drug Discovery and Protein Science]()
AlphaFold 3 and Protein Structure Prediction: Transforming Drug Discovery and Protein Science
bioinformatics![Spatial Metabolomics]()
Introduction to Quantitative Proteomics: An Advanced Outline
proteomics![proteomics-omics]()
How Proteomics is Contributing to Vaccine Development
proteomics![proteomics-omics]()
Proteogenomics
proteomics![Single-Cell-Proteomics]()
Introduction to Single-Cell Proteomics
proteomics![Proteomics]()
10 Breakthroughs in Proteomics Everyone Should Know About
proteomics![Proteomics]()
What is Proteomics
proteomics![A Submitter's Guide to GenBank]()
A Submitter's Guide to GenBank
Guides![proteomics]()
Protein Identification Methods in Proteomics
Guides![Spatial-Proteomics]()
Essential Imaging Software for Mass Spectrometry and Molecular Visualization
bioinformatics![proteomics-quantative]()
Quantitative Proteomics Techniques
proteomics![3Dstructureofprotein-deepmind]()
Deep Learning in Drug Discovery: Predicting Protein-Ligand Binding Affinity
A.I![proteomics]()
Latest Breakthroughs in Targeted Protein Degradation
proteomics