Introduction to Neuroproteomics in Neurodegenerative Diseases
February 16, 2024Introduction
Neuroproteomics is the study of the proteome of the nervous system, including the brain, spinal cord, and peripheral nerves. It involves the comprehensive analysis of the proteins expressed in these tissues to understand their functions, interactions, and alterations in disease states. Neuroproteomics plays a crucial role in advancing our understanding of neurodegenerative diseases, such as Alzheimer’s disease and Parkinson’s disease, by elucidating the molecular mechanisms underlying these disorders.
One of the key aspects of neuroproteomics is the study of protein misfolding and aggregation, which are hallmark features of many neurodegenerative diseases. Misfolded proteins can form aggregates that are toxic to neurons and disrupt normal cellular functions, leading to neuronal dysfunction and cell death. Understanding the mechanisms underlying protein misfolding and aggregation is essential for developing effective therapies for these devastating diseases.
Alzheimer’s disease is characterized by the accumulation of misfolded amyloid-beta and tau proteins in the brain, leading to the formation of plaques and tangles, respectively. Parkinson’s disease is characterized by the accumulation of misfolded alpha-synuclein protein in neurons, forming Lewy bodies. Studying the proteomic changes associated with these diseases can provide insights into their pathogenesis and identify potential therapeutic targets.
In summary, neuroproteomics is a powerful tool for studying the molecular mechanisms of neurodegenerative diseases, with the potential to uncover novel biomarkers and therapeutic targets. Understanding protein misfolding and aggregation in these diseases is crucial for developing effective treatments and ultimately improving the lives of patients affected by these devastating disorders.
Neurodegenerative Diseases: Overview
Neurodegenerative diseases are a group of disorders characterized by the progressive degeneration of neurons in the brain and/or spinal cord. These diseases are typically chronic and debilitating, leading to gradual loss of cognitive function, motor control, and in some cases, autonomy. Some of the most common neurodegenerative diseases include Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and amyotrophic lateral sclerosis (ALS).
A common feature of many neurodegenerative diseases is the abnormal folding and aggregation of specific proteins in the brain. These misfolded proteins can accumulate into insoluble aggregates, such as plaques and tangles, which are toxic to neurons and disrupt normal cellular function. This process of protein misfolding and aggregation is thought to play a central role in the pathogenesis of these diseases.
The impact of neurodegenerative diseases on patients and healthcare systems is profound. Patients with these diseases often experience a gradual decline in cognitive function, memory loss, impaired movement, and other neurological symptoms. These symptoms can significantly reduce quality of life and independence, requiring extensive care and support from healthcare providers and caregivers.
From a healthcare perspective, neurodegenerative diseases pose significant challenges due to their chronic and progressive nature. They often require long-term management and care, placing a substantial burden on healthcare systems and resources. Additionally, the lack of effective disease-modifying treatments for many neurodegenerative diseases further complicates their management and adds to the economic burden of these conditions.
In summary, neurodegenerative diseases are characterized by progressive neuronal degeneration, protein misfolding, and aggregation, leading to significant impact on patients’ lives and healthcare systems. Understanding the underlying mechanisms of these diseases is crucial for developing effective treatments and improving patient outcomes.
Protein Misfolding and Aggregation in Neurodegenerative Diseases
Mechanisms of Protein Misfolding and Aggregation
1. Protein Misfolding: Proteins are synthesized as linear chains of amino acids and must fold into specific three-dimensional structures to function properly. Misfolding can occur due to genetic mutations, environmental factors, or errors in protein folding machinery.
2. Aggregation: Misfolded proteins have a tendency to aggregate, forming oligomers and eventually insoluble fibrils. This process is often facilitated by interactions between exposed hydrophobic regions of misfolded proteins.
Role of Misfolded Proteins in Disease Progression
1. Disruption of Cellular Function: Aggregated proteins can interfere with normal cellular processes, such as protein degradation and transport, leading to cellular dysfunction and eventually cell death.
2. Formation of Inclusions: Aggregated proteins can form intracellular or extracellular inclusions, such as Lewy bodies in Parkinson’s disease or amyloid plaques in Alzheimer’s disease, which are characteristic pathological features of these diseases.
Relationship Between Protein Aggregation and Neurotoxicity
1. Oxidative Stress: Aggregated proteins can induce oxidative stress, leading to damage to cellular structures and DNA, and ultimately cell death.
2. Mitochondrial Dysfunction: Aggregated proteins can disrupt mitochondrial function, leading to energy depletion and increased production of reactive oxygen species (ROS), contributing to neuronal damage.
3. Activation of Immune Response: Aggregated proteins can activate microglia and astrocytes, leading to neuroinflammation, which can exacerbate neuronal damage.
In summary, protein misfolding and aggregation play a central role in the pathogenesis of neurodegenerative diseases. Misfolded proteins can disrupt cellular function, form toxic aggregates, and induce neurotoxicity, ultimately leading to neuronal dysfunction and cell death. Understanding the mechanisms underlying protein aggregation is crucial for developing effective therapeutic strategies for these devastating diseases.
Neuroproteomics Approaches
Techniques for Studying Protein Misfolding and Aggregation
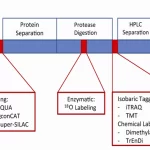
1. Mass Spectrometry: Mass spectrometry can be used to identify and quantify proteins in complex samples, such as brain tissue or cerebrospinal fluid, allowing researchers to analyze the presence and abundance of misfolded or aggregated proteins.
2. Gel Electrophoresis: Gel electrophoresis techniques, such as SDS-PAGE and native PAGE, can be used to separate proteins based on size and charge. This allows researchers to visualize protein aggregates and analyze their composition.
3. Immunohistochemistry: Immunohistochemistry involves using antibodies to detect specific proteins in tissue samples. It can be used to visualize the distribution of misfolded or aggregated proteins in the brain and other tissues.
Application of Neuroproteomics in Identifying Biomarkers and Potential Drug Targets
1. Biomarker Discovery: Neuroproteomics can be used to identify protein biomarkers associated with neurodegenerative diseases. These biomarkers can be used for early diagnosis, monitoring disease progression, and assessing treatment response.
2. Drug Target Identification: By studying the proteomic changes associated with neurodegenerative diseases, researchers can identify potential drug targets. Targeted therapies aimed at these proteins could help prevent or slow down disease progression.
3. Mechanistic Studies: Neuroproteomics can provide insights into the molecular mechanisms underlying protein misfolding and aggregation in neurodegenerative diseases. This knowledge is essential for developing targeted therapies that can effectively intervene in disease progression.
In summary, techniques such as mass spectrometry, gel electrophoresis, and immunohistochemistry are valuable tools for studying protein misfolding and aggregation in neurodegenerative diseases. Neuroproteomics has the potential to identify biomarkers for early diagnosis and treatment monitoring, as well as to identify potential drug targets for the development of novel therapies.
Alzheimer’s Disease: A Tauopathy
Alzheimer’s disease is characterized by the abnormal accumulation of two types of protein aggregates in the brain: amyloid-beta plaques and neurofibrillary tangles (NFTs). NFTs are primarily composed of hyperphosphorylated tau protein, leading to the classification of Alzheimer’s disease as a tauopathy.
The tau protein is normally involved in stabilizing microtubules, which are essential for neuronal structure and function. In Alzheimer’s disease, tau becomes hyperphosphorylated, causing it to detach from microtubules and aggregate into insoluble filaments, forming NFTs. These NFTs disrupt the normal functioning of neurons, leading to neuronal dysfunction and eventually cell death.
Neuroproteomics studies on tau protein in Alzheimer’s disease have focused on understanding the post-translational modifications, interactions, and aggregation patterns of tau. Mass spectrometry-based approaches have been used to identify specific phosphorylation sites on tau and to quantify the levels of hyperphosphorylated tau in different brain regions and disease stages. These studies have provided insights into the molecular mechanisms underlying tau pathology in Alzheimer’s disease and have identified potential biomarkers and therapeutic targets for the disease.
Parkinson’s Disease: A Synucleinopathy
Parkinson’s disease (PD) is a neurodegenerative disorder characterized by the loss of dopaminergic neurons in the substantia nigra region of the brain. It is primarily known for its motor symptoms, including tremors, rigidity, and bradykinesia. PD is also associated with non-motor symptoms, such as cognitive impairment and autonomic dysfunction.
PD is considered a synucleinopathy because it is characterized by the accumulation of alpha-synuclein protein aggregates, known as Lewy bodies, in affected neurons. Alpha-synuclein is a presynaptic protein that normally plays a role in regulating synaptic vesicle function and neurotransmitter release. In PD, alpha-synuclein misfolds and aggregates into insoluble fibrils, which accumulate in neurons and disrupt normal cellular function.
Neuroproteomics studies on alpha-synuclein in PD have focused on understanding the role of alpha-synuclein in disease pathogenesis, as well as identifying potential biomarkers and therapeutic targets. Mass spectrometry-based approaches have been used to analyze the post-translational modifications, interactions, and aggregation patterns of alpha-synuclein in PD brains. These studies have provided insights into the molecular mechanisms underlying alpha-synuclein pathology and have identified potential targets for drug development and biomarkers for disease diagnosis and progression.
Other Neurodegenerative Diseases
In addition to Alzheimer’s disease and Parkinson’s disease, there are several other neurodegenerative diseases characterized by protein misfolding and aggregation. Two prominent examples are Huntington’s disease (HD) and amyotrophic lateral sclerosis (ALS).
Huntington’s Disease (HD): HD is a genetic neurodegenerative disorder caused by an expansion of CAG repeats in the huntingtin (HTT) gene. The mutant huntingtin protein (mHTT) produced from this gene contains an abnormally long polyglutamine tract, which leads to protein misfolding and aggregation. The aggregates of mHTT accumulate in neurons, particularly in the striatum, leading to neuronal dysfunction and cell death. HD is characterized by motor abnormalities, cognitive decline, and psychiatric symptoms.
Amyotrophic Lateral Sclerosis (ALS): ALS is a progressive neurodegenerative disease that affects motor neurons in the brain and spinal cord. While most cases of ALS are sporadic, about 10% are familial and associated with mutations in genes such as C9orf72, SOD1, and TARDBP. In ALS, misfolded proteins, including mutant forms of superoxide dismutase 1 (SOD1) and TAR DNA-binding protein 43 (TDP-43), form aggregates in motor neurons. These aggregates disrupt cellular function and lead to the degeneration of motor neurons, resulting in muscle weakness, paralysis, and eventually respiratory failure.
Other Examples:
- Prion Diseases: Prion diseases, such as Creutzfeldt-Jakob disease (CJD) and mad cow disease, are caused by misfolded prion proteins that can induce the misfolding of normal prion proteins, leading to the accumulation of prion aggregates in the brain.
- Spinocerebellar Ataxias (SCAs): SCAs are a group of genetic disorders characterized by progressive cerebellar ataxia. Several SCAs are caused by CAG repeat expansions in specific genes, leading to the production of mutant proteins that misfold and form aggregates in neurons.
- Frontotemporal Dementia (FTD): FTD is a neurodegenerative disorder characterized by progressive degeneration of the frontal and temporal lobes of the brain. Some forms of FTD are associated with mutations in genes such as MAPT (encoding tau protein) and C9orf72, leading to protein misfolding and aggregation.
In summary, several neurodegenerative diseases are characterized by protein misfolding and aggregation, which contribute to neuronal dysfunction and cell death. Understanding the mechanisms underlying protein misfolding and aggregation in these diseases is crucial for developing effective treatments.
Challenges and Future Directions
Challenges in Studying Protein Misfolding and Aggregation
- Complexity of Protein Interactions: Proteins involved in neurodegenerative diseases often interact with multiple partners, making it challenging to decipher the specific interactions that lead to misfolding and aggregation.
- Temporal and Spatial Dynamics: Misfolding and aggregation processes can vary between different stages of disease and in different regions of the brain, requiring spatially and temporally resolved studies.
- Detection Sensitivity: Protein aggregates can be present in low concentrations, requiring highly sensitive detection methods for accurate quantification and characterization.
- Sample Heterogeneity: Biological samples, such as brain tissue or cerebrospinal fluid, are heterogeneous, requiring careful sample preparation and analysis to account for variability.
- Limited Access to Human Samples: Access to human brain tissue samples, especially from early stages of disease, is limited, making it challenging to study disease progression.
Future Directions in Neuroproteomics Research
- Single-Cell Proteomics: Advancements in single-cell proteomics can help elucidate cell-specific proteomic changes in neurodegenerative diseases, providing insights into cellular heterogeneity and disease progression.
- Spatial Proteomics: Techniques such as spatial proteomics can provide information on the subcellular localization of proteins and aggregates, helping to understand their impact on cellular function.
- Longitudinal Studies: Longitudinal studies in animal models and patient cohorts can provide insights into the temporal dynamics of protein misfolding and aggregation in neurodegenerative diseases.
- Multi-Omic Integration: Integrating proteomic data with other omic datasets, such as genomic and transcriptomic data, can provide a comprehensive understanding of disease mechanisms.
- Machine Learning and AI: Utilizing machine learning and AI algorithms can help analyze large-scale proteomic datasets, identify disease signatures, and predict disease progression.
Potential Therapeutic Strategies Targeting Protein Misfolding and Aggregation
- Targeted Protein Degradation: Developing molecules that target and degrade misfolded proteins, such as proteasome inhibitors or autophagy inducers, to reduce protein aggregation.
- Stabilizing Protein Structure: Small molecules or antibodies that stabilize the native conformation of proteins, preventing them from misfolding and aggregating.
- Immunotherapy: Using antibodies to target and clear misfolded proteins, such as beta-amyloid or alpha-synuclein, from the brain.
- Chaperone Therapies: Molecular chaperones that assist in protein folding and prevent misfolding and aggregation.
- Gene Therapy: Targeting the expression of genes involved in protein misfolding and aggregation, such as using RNA interference to reduce the expression of mutant proteins.
In conclusion, studying protein misfolding and aggregation in neurodegenerative diseases presents several challenges, but advancements in neuroproteomics research hold promise for understanding disease mechanisms and developing effective therapeutic strategies.
Conclusion
Neuroproteomics plays a crucial role in unraveling the complexities of protein misfolding and aggregation in neurodegenerative diseases. By studying the post-translational modifications, interactions, and aggregation patterns of key proteins such as tau and alpha-synuclein, neuroproteomics helps identify biomarkers, understand disease mechanisms, and develop potential therapeutic strategies. However, challenges such as sample heterogeneity and limited access to human samples remain, highlighting the need for continued research and collaboration in the field of neuroproteomics. By addressing these challenges and leveraging advancements in technology and data analysis, neuroproteomics holds great promise for advancing our understanding of neurodegenerative diseases and developing effective treatments.
Related posts:
![Mass Spectrometry in Proteomics: A Comprehensive Guide for 2023]()
Mass Spectrometry in Proteomics: A Comprehensive Guide for 2023
Guides![Proteomics]()
10 Breakthroughs in Proteomics Everyone Should Know About
proteomics![AlphaFold 3 and Protein Structure Prediction: Transforming Drug Discovery and Protein Science]()
AlphaFold 3 and Protein Structure Prediction: Transforming Drug Discovery and Protein Science
bioinformatics![spatialtranscriptomics]()
Spatial Omics: Revolutionizing Tissue-Level Molecular Insights
proteomics![3Dstructureofprotein-deepmind]()
Deep Learning in Drug Discovery: Predicting Protein-Ligand Binding Affinity
A.I![Humangenomeproject-personalizedmedicine]()
Personalized Medicine and Proteomics: The Perfect Pair
proteomics![proteomics-omics]()
Unveiling the Synergy of Proteomics Data in Bioinformatics: Challenges and Successes in Multi-Omics ...
Multiomics![Proteomics]()
What is Proteomics
proteomics![Omics data analysis]()
Essential Checklist for Integrating Omics Data: What You Need to Know for Effective Analysis
bioinformatics![Deepmind-Aplhafold]()
Protein Structure Determination and Prediction
proteomics![Proteomics tools]()
How are proteomics datasets analyzed to reveal protein regulation?
proteomics![Neuroproteomics]()
Proteomics: Methods & Applications
Guides![proteomics]()
Demystifying Proteomics: The Promise and Challenges of Protein Sequencing
proteomics![A Submitter's Guide to GenBank]()
A Submitter's Guide to GenBank
Guides![proteomics-quantative]()
Quantitative Proteomics Techniques
proteomics![Protein Separation Techniques in Proteomics]()
Protein Separation Techniques in Proteomics
Guides