Introduction to Quantitative Proteomics: An Advanced Outline
February 16, 2024Introduction
Definition of Quantitative Proteomics
Quantitative proteomics is a branch of proteomics that focuses on the large-scale study of the expression levels of proteins in biological samples. It involves the identification and quantification of proteins to understand their roles in various biological processes. Quantitative proteomics aims to measure changes in protein expression levels, post-translational modifications, protein-protein interactions, and protein turnover rates in response to different conditions or stimuli.
Quantitative proteomics techniques often involve the use of mass spectrometry to identify and quantify proteins in complex biological samples. These techniques can provide insights into the dynamics of protein expression and function, helping researchers understand the molecular mechanisms underlying various biological processes, including disease development and progression.
Importance of accurately measuring protein abundance and dynamics in understanding cellular processes
Accurately measuring protein abundance and dynamics is crucial for understanding cellular processes for several reasons:
- Functional Insights: Proteins are the primary effectors of cellular functions. Changes in protein abundance and dynamics directly impact cellular processes such as signaling, metabolism, and gene regulation. Quantitative proteomics allows researchers to link changes in protein levels to specific cellular functions, providing insights into the molecular mechanisms underlying biological processes.
- Disease Mechanisms: Aberrant protein expression and dynamics are often associated with disease. Quantitative proteomics can identify disease-specific changes in protein abundance and post-translational modifications, leading to the discovery of potential biomarkers and therapeutic targets.
- Dynamic Responses: Cells respond to environmental cues and stimuli by modulating protein expression and activity. Quantitative proteomics enables the study of dynamic changes in protein abundance and modifications in response to different conditions, providing insights into cellular adaptation and response mechanisms.
- Protein Interactions: Proteins rarely act alone but often interact with other proteins to form complexes. Quantitative proteomics can identify and quantify protein-protein interactions, helping researchers understand the architecture and dynamics of protein complexes involved in cellular processes.
- Drug Discovery: Understanding protein abundance and dynamics is critical for drug discovery and development. Quantitative proteomics can identify proteins that are dysregulated in disease and assess the effects of drugs on protein expression and activity, aiding in the development of targeted therapies.
In summary, accurately measuring protein abundance and dynamics is essential for gaining a comprehensive understanding of cellular processes, disease mechanisms, and drug responses. Quantitative proteomics provides a powerful tool for studying protein expression and function, leading to advances in basic biological research and clinical applications.
Overview of key technologies and concepts: label-free quantitative proteomics, stable isotope labeling, absolute protein quantification
Label-Free Quantitative Proteomics
Definition: Label-free quantitative proteomics is a method for quantifying proteins based on the comparison of peptide signal intensities between different samples, without the use of chemical or isotopic labels.
Key Concepts:
- Peptide Signal Intensity: The intensity of peptide signals in mass spectrometry data is used as a measure of protein abundance.
- Data Normalization: To account for technical variation, data normalization methods are used to adjust peptide intensities between samples.
- Data Analysis: Statistical methods are applied to identify significant differences in protein abundance between samples and to infer biological insights.
Advantages:
- Simplicity and cost-effectiveness compared to labeled methods.
- Suitable for analyzing large sample cohorts and complex experimental designs.
- Limited dynamic range and sensitivity compared to labeled methods.
- Susceptibility to variability in sample preparation and data analysis.
Stable Isotope Labeling
Definition: Stable isotope labeling is a method for quantifying proteins by incorporating stable isotopes into peptides or proteins, allowing for accurate quantification using mass spectrometry.
Key Concepts:
- Isotopic Labeling: Isotopes such as ^2H, ^13C, or ^15N are incorporated into proteins or peptides either metabolically (in vivo) or chemically (in vitro).
- Isotope Ratio Measurement: Mass spectrometry is used to measure the ratio of isotopically labeled peptides to unlabeled peptides, allowing for quantification.
- Quantification Accuracy: Stable isotope labeling provides accurate quantification and is suitable for both relative and absolute quantification.
Advantages:
- High accuracy and precision in quantification.
- Suitable for dynamic range and low-abundance protein detection.
Limitations:
- More complex sample preparation and data analysis compared to label-free methods.
- Higher cost and requirement for specialized reagents.
Absolute Protein Quantification
Definition: Absolute protein quantification is a method for determining the absolute concentration of proteins in a sample, typically using mass spectrometry-based techniques.
Key Concepts:
- Internal Standards: Isotopically labeled peptide standards are used to generate calibration curves for absolute quantification.
- Standard Addition Method: Known amounts of isotopically labeled peptides are added to the sample, and the ratio of labeled to unlabeled peptides is used to calculate absolute concentrations.
- Protein Copy Number Determination: Absolute protein quantification can provide insights into the stoichiometry of protein complexes and the absolute abundance of proteins in cells.
Advantages:
- Provides quantitative information on protein copy numbers and absolute concentrations.
- Useful for studying protein stoichiometry and cellular abundance.
Limitations:
- Requires careful calibration and validation of standards.
- More technically challenging and time-consuming than relative quantification methods.
In summary, label-free quantitative proteomics, stable isotope labeling, and absolute protein quantification are key technologies used in quantitative proteomics for studying protein abundance and dynamics. Each method has its advantages and limitations, and the choice of method depends on the research question and experimental design.
Principles of Quantitative Proteomics
Protein Quantification Methods: Label-Based vs. Label-Free Approaches
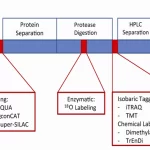
Label-Based Approaches:
- Definition: Label-based approaches involve introducing a chemical or isotopic label into proteins or peptides to distinguish between different samples.
- Principle: The label is incorporated into proteins or peptides either metabolically or chemically, allowing for accurate quantification using mass spectrometry.
- Examples: Stable isotope labeling by amino acids in cell culture (SILAC), isobaric tags for relative and absolute quantification (iTRAQ), tandem mass tags (TMT).
Label-Free Approaches:
- Definition: Label-free approaches quantify proteins based on the comparison of peptide signal intensities between different samples, without the use of chemical or isotopic labels.
- Principle: Protein abundance is inferred from the intensity of peptide signals in mass spectrometry data, with normalization applied to correct for technical variation.
- Examples: Spectral counting, intensity-based absolute quantification (iBAQ).
Principles of Label-Free Quantitative Proteomics
- Measurement: Label-free quantification relies on the comparison of peptide signal intensities between different samples.
- Normalization: To correct for technical variability, data normalization methods are applied to adjust peptide intensities.
- Data Analysis: Statistical methods are used to identify significant differences in protein abundance between samples and to infer biological insights.
- Advantages: Label-free quantification is simple, cost-effective, and suitable for analyzing large sample cohorts and complex experimental designs.
- Limitations: It has a limited dynamic range and sensitivity compared to labeled methods and is susceptible to variability in sample preparation and data analysis.
Role of Stable Isotope Labeling in Quantifying Protein Abundance
- Accurate Quantification: Stable isotope labeling provides accurate quantification by incorporating isotopes into peptides or proteins, allowing for precise measurement using mass spectrometry.
- Dynamic Range: Stable isotope labeling enables the detection and quantification of proteins across a wide dynamic range, including low-abundance proteins.
- Absolute Quantification: Stable isotope labeling can be used for absolute quantification by using isotopically labeled peptide standards or the standard addition method.
- Applications: Stable isotope labeling is used in various quantitative proteomics studies, such as studying protein dynamics, post-translational modifications, and protein-protein interactions.
In summary, both label-based and label-free approaches are valuable methods for quantifying protein abundance in quantitative proteomics. Label-free approaches are simple and cost-effective but have limitations in dynamic range and sensitivity. Stable isotope labeling provides accurate and absolute quantification, making it suitable for a wide range of quantitative proteomics applications.
Technologies in Quantitative Proteomics
Label-Free Quantitative Proteomics: Workflow and Data Analysis
Workflow:
- Sample Preparation: Proteins are extracted from cells or tissues and digested into peptides.
- LC-MS/MS Analysis: Peptides are separated using liquid chromatography (LC) and analyzed by tandem mass spectrometry (MS/MS).
- Data Acquisition: Mass spectrometry data is acquired, and peptide signals are recorded.
- Data Processing: Peptide signals are quantified, and data normalization is performed to correct for technical variation.
- Statistical Analysis: Statistical methods are applied to identify significantly changed proteins between samples and to infer biological insights.
Data Analysis:
- Label-Free Quantification: Protein abundance is inferred from the intensity of peptide signals in mass spectrometry data.
- Normalization: Data normalization methods such as total peptide amount normalization or internal standard normalization are applied to correct for technical variability.
- Statistical Analysis: Statistical tests, such as t-tests or ANOVA, are used to identify proteins that are significantly changed between samples.
- Biological Interpretation: Identified proteins are analyzed for their biological functions and pathways to understand their roles in cellular processes.
Stable Isotope Labeling Techniques
- SILAC (Stable Isotope Labeling by Amino Acids in Cell Culture):
- Principle: Cells are cultured in medium containing isotopically labeled amino acids (e.g., heavy lysine or arginine), leading to the incorporation of labels into newly synthesized proteins.
- Quantification: Proteins from labeled and unlabeled cells are mixed, and the ratio of labeled to unlabeled peptides is measured by mass spectrometry for quantification.
- TMT (Tandem Mass Tagging):
- Principle: Peptide samples are labeled with isobaric tags that contain different isotopic compositions, allowing for multiplexed quantification of up to 10 samples in a single mass spectrometry analysis.
- Quantification: The intensity of reporter ions in the mass spectra is used to quantify peptides in each sample.
- iTRAQ (Isobaric Tags for Relative and Absolute Quantitation):
- Principle: Peptide samples are labeled with isobaric tags that contain different isotopic compositions, allowing for multiplexed quantification of up to 8 samples in a single mass spectrometry analysis.
- Quantification: Similar to TMT, the intensity of reporter ions in the mass spectra is used to quantify peptides in each sample.
Absolute Protein Quantification Methods
- AQUA (Absolute QUAntification) Peptides:
- Principle: Isotopically labeled peptide standards are synthesized with known concentrations and used as internal standards for absolute quantification.
- Quantification: The ratio of labeled to unlabeled peptides is measured by mass spectrometry to determine the absolute concentration of the target protein.
- SRM (Selected Reaction Monitoring):
- Principle: Specific peptide ions are selected for monitoring based on their mass-to-charge ratio (m/z) and fragmentation pattern.
- Quantification: The intensity of selected peptide ions is measured over time to quantify the target protein.
These techniques play a crucial role in quantitative proteomics by providing accurate and reliable quantification of proteins in complex biological samples.
Applications of Quantitative Proteomics
Quantitative Analysis of Protein Expression Changes in Response to Stimuli or Treatments
Objective: To quantify changes in protein expression levels in response to stimuli or treatments, providing insights into cellular responses and signaling pathways.
Workflow:
- Experimental Design: Samples are treated with different stimuli or treatments, and control samples are included for comparison.
- Protein Extraction and Digestion: Proteins are extracted from samples, digested into peptides, and labeled (if applicable).
- Mass Spectrometry Analysis: Peptides are separated by liquid chromatography and analyzed by mass spectrometry to quantify protein expression levels.
- Data Analysis: Quantitative proteomics software is used to compare protein expression levels between treated and control samples, identifying significantly changed proteins.
Biological Insights:
- Identification of proteins and pathways involved in the cellular response to stimuli or treatments.
- Understanding the mechanisms underlying biological processes, such as cell signaling and regulation.
Dynamics of Protein-Protein Interactions and Post-Translational Modifications
Objective: To study the dynamic interactions between proteins and the regulation of protein function through post-translational modifications (PTMs).
Workflow:
- Protein Complex Isolation: Protein complexes are isolated from cells or tissues using affinity purification techniques.
- Cross-Linking (Optional): Proteins within complexes can be cross-linked to preserve transient interactions.
- Protein Digestion: Proteins are digested into peptides, and PTMs are preserved.
- Mass Spectrometry Analysis: Peptides are analyzed by mass spectrometry to identify protein-protein interactions and PTMs.
- Quantitative Analysis: Quantitative proteomics methods are used to quantify changes in protein interactions and PTMs under different conditions.
Biological Insights:
- Mapping of protein interaction networks and signaling pathways.
- Understanding the role of PTMs in regulating protein function and cellular processes.
Biomarker Discovery and Validation in Disease Research
Objective: To identify and validate protein biomarkers for disease diagnosis, prognosis, and treatment response.
Workflow:
- Clinical Sample Collection: Blood, tissue, or other clinical samples are collected from patients with and without the disease of interest.
- Protein Extraction and Analysis: Proteins are extracted from samples and analyzed using quantitative proteomics techniques to identify candidate biomarkers.
- Validation Studies: Candidate biomarkers are validated in independent patient cohorts using targeted proteomics assays, such as SRM or immunoassays.
- Clinical Translation: Validated biomarkers are further evaluated for their clinical utility and potential applications in disease diagnosis, prognosis, and treatment monitoring.
Biological Insights:
- Discovery of novel biomarkers for early disease detection, patient stratification, and treatment response prediction.
- Understanding the molecular mechanisms underlying disease pathogenesis and progression.
In summary, quantitative proteomics enables the study of protein expression changes, protein-protein interactions, and post-translational modifications in response to stimuli or treatments, as well as the discovery and validation of biomarkers for disease research. These applications provide valuable insights into biological processes and disease mechanisms, with potential clinical implications for diagnosis, prognosis, and treatment.
Label-Free Quantitative Proteomics
Related posts:
![Proteomics tools]()
Overview of Recent Advancements in Proteomics Bioinformatics Tools
proteomics![proteomics-omics]()
Protein Structures and Post-translational Modifications: An Overview
proteomics![proteomics-quantative]()
Quantitative Proteomics Techniques
proteomics![AlphaFold 3 and Protein Structure Prediction: Transforming Drug Discovery and Protein Science]()
AlphaFold 3 and Protein Structure Prediction: Transforming Drug Discovery and Protein Science
bioinformatics![Spatial-Proteomics]()
Introduction to Spatial Proteomics
proteomics![Single-Cell-Proteomics]()
Introduction to Single-Cell Proteomics
proteomics![Humangenomeproject-personalizedmedicine]()
Personalized Medicine and Proteomics: The Perfect Pair
proteomics![Proteomics]()
10 Breakthroughs in Proteomics Everyone Should Know About
proteomics![Global Genomics Initiatives]()
Proteogenomics: Integrating Proteomics with Genomics
genomics![proteomics-omics]()
Unveiling the Synergy of Proteomics Data in Bioinformatics: Challenges and Successes in Multi-Omics ...
Multiomics![Proteomics-Quick-Study-A-Brief-Introduction]()
Proteomics Quick Study: A Brief Introduction
Guides![next-generation-sequencing-technology]()
Generative AI in De Novo Drug Design
A.I![A Submitter's Guide to GenBank]()
A Submitter's Guide to GenBank
Guides![proteomics-omics]()
Unlock Deeper Proteomics Insights with Advanced Mass Spectrometry
proteomics![proteome]()
Proteomics and Biomarker Discovery: Using Proteomics to Find New Disease Biomarkers
proteomics![Proteomics]()
What is Proteomics
proteomics