How are proteomics datasets analyzed to reveal protein regulation?
November 24, 2023I. Introduction
A. Definition of Proteomics: Proteomics is the comprehensive study of the entire set of proteins within a biological system, including their structures, functions, interactions, and modifications. It involves the systematic analysis of proteins to understand their roles in cellular processes and broader biological phenomena.
B. Importance of Proteomics in Understanding Cellular Processes:
- Holistic View of Cell Function: Proteomics provides insights into the dynamic and functional aspects of cellular processes by examining the complete protein complement.
- Identification of Biomolecules: It aids in identifying and characterizing proteins, enabling the discovery of potential biomarkers for various physiological and pathological conditions.
- Protein-Protein Interactions: Proteomics helps elucidate the intricate network of protein-protein interactions, crucial for understanding signaling pathways and cellular regulation.
- Post-Translational Modifications (PTMs): By studying PTMs, such as phosphorylation and glycosylation, proteomics reveals how proteins are modified, influencing their activities and cellular localization.
- Drug Discovery: Understanding the proteome is fundamental in drug development, as many drugs target specific proteins. Proteomics aids in identifying potential drug targets and assessing treatment responses.
C. Overview of Protein Regulation Analysis: Protein regulation analysis in proteomics involves investigating the factors influencing protein expression, localization, and modifications. This includes studying the impact of environmental cues, cellular signaling pathways, and genetic variations on the proteome. The integration of proteomic data with other omics disciplines contributes to a comprehensive understanding of biological systems.
II. Types of Proteomics Datasets
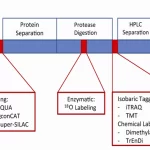
A. Mass Spectrometry Data:
- Identification of Proteins: Mass spectrometry is a pivotal tool for identifying proteins within a sample. It analyzes the mass-to-charge ratio of ionized proteins, allowing for the determination of their identity.
- Quantification of Protein Abundance: Mass spectrometry facilitates the quantification of protein abundance. Techniques like label-free or isotope labeling methods enable the comparison of protein levels across different conditions.
B. Protein-Protein Interaction Networks:
- Studying Protein Connectivity: Proteomics contributes to the construction of protein-protein interaction networks. By identifying interacting partners, researchers can elucidate the complex web of connections between proteins.
- Insights into Functional Relationships: Analyzing protein-protein interaction networks provides insights into the functional relationships between proteins. It helps discern modules or pathways within the network, revealing coordinated cellular activities.
III. Preprocessing and Data Cleaning
A. Raw Data Processing:
- Conversion of Raw Mass Spectrometry Data: Raw mass spectrometry data undergoes processing to convert complex signals into interpretable formats. This step involves transforming raw spectra into lists of detected ions and their intensities.
- Quality Control and Normalization: To ensure data quality and comparability, quality control measures are applied. Normalization techniques are used to correct for systematic variations, providing a reliable basis for quantitative analysis.
B. Missing Value Imputation:
- Addressing Missing Data in Proteomics Datasets: Proteomics datasets often contain missing values due to technical challenges or inherent variability. Strategies are employed to handle missing data, preventing biases in downstream analyses.
- Techniques for Imputing Missing Values: Various imputation methods, such as mean imputation, k-nearest neighbors, or advanced statistical approaches, are applied to estimate missing values accurately and maintain the integrity of the dataset.
IV. Differential Expression Analysis
A. Quantitative Comparison of Protein Abundance:
- Identification of Differentially Expressed Proteins: Differential expression analysis aims to identify proteins whose abundance significantly differs between experimental conditions. This involves comparing quantitative measures, such as spectral counts or intensities, across samples.
- Statistical Methods for Significance Testing: Various statistical tests, such as t-tests, ANOVA, or more advanced methods like edgeR or DESeq2, are employed to assess the significance of observed differences and determine differentially expressed proteins.
B. Volcano Plots and Heatmaps:
- Visual Representation of Differential Expression: Volcano plots graphically represent fold change against statistical significance, aiding in the visualization of differentially expressed proteins. Proteins with significant changes are positioned away from the center.
- Clustering Analysis for Pattern Recognition: Heatmaps provide a visual summary of protein expression patterns. Clustering algorithms group proteins with similar expression profiles, revealing potential biological insights and relationships between experimental conditions.
V. Pathway and Functional Enrichment Analysis
A. Mapping Proteins to Biological Pathways:
- Identifying Pathways Enriched with Regulated Proteins: After differential expression analysis, proteins are often mapped to biological pathways using pathway enrichment analysis. This helps identify pathways significantly enriched with proteins that show altered abundance.
- Tools for Pathway Analysis: Various tools, such as DAVID, Reactome, or KEGG, are commonly used to perform pathway enrichment analysis and gain insights into the biological processes affected by changes in protein abundance.
B. Gene Ontology Analysis:
- Categorizing Proteins Based on Biological Processes: Gene Ontology (GO) analysis categorizes proteins into biological processes, molecular functions, and cellular components. This systematic classification provides a broad overview of the functional roles of differentially expressed proteins.
- Functional Annotation of Regulated Proteins: Understanding the functional annotations of proteins aids in deciphering the impact of differential expression on cellular functions. GO analysis helps in attributing biological significance to the observed changes in protein abundance.
VI. Post-translational Modification (PTM) Analysis
A. Identification of PTMs:
- Phosphorylation, Acetylation, Glycosylation, etc.: Proteins undergo various post-translational modifications (PTMs), such as phosphorylation, acetylation, glycosylation, etc. These modifications play crucial roles in regulating protein function, localization, and interactions within cellular processes.
- Impact on Protein Function and Regulation: Understanding the specific PTMs occurring on proteins provides insights into their functional consequences. For example, phosphorylation can regulate enzymatic activity, while acetylation may impact protein stability and gene expression.
B. PTM Site Localization:
- Tools for Accurate Site Localization: Mass spectrometry-based techniques, including tandem mass spectrometry (MS/MS), are commonly used for identifying PTM sites. Specialized bioinformatics tools, such as MaxQuant and Mascot, aid in the accurate identification and site localization of PTMs.
- Functional Implications of Specific PTMs: Knowing the exact sites of PTMs is crucial for understanding their functional implications. Site-specific PTMs can have distinct roles in cellular processes, and analyzing their functional consequences enhances the comprehension of protein regulation and signaling pathways.
VII. Protein-Protein Interaction Network Analysis
A. Construction of Interaction Networks:
- Data Integration for Network Creation: Protein-protein interaction (PPI) networks are constructed by integrating experimental data from methods like yeast two-hybrid assays, co-immunoprecipitation, and computational predictions. Tools like STRING and BioGRID facilitate data compilation for network construction.
- Visualization of Protein-Protein Interactions: Network visualization tools, such as Cytoscape, enable the representation and exploration of PPI networks. Visualization aids in identifying patterns, clusters, and central nodes within the interaction network.
B. Network Metrics and Analysis:
- Identifying Hub Proteins: Network metrics, including degree centrality, betweenness centrality, and closeness centrality, help identify hub proteins. Hubs are highly connected nodes that play key roles in maintaining network integrity and are often crucial for cellular functions.
- Modules and Clusters in Interaction Networks: Analyzing PPI networks reveals functional modules and clusters, representing groups of proteins with interconnected roles. Detection of modules enhances the understanding of protein cooperation in specific pathways or cellular processes. Tools like MCODE assist in module identification.
VIII. Integration with Other Omics Data
A. Correlation with Transcriptomics Data:
- Cross-referencing Proteomics Findings with Gene Expression: Integrating proteomics with transcriptomics data allows the correlation of protein abundance with corresponding mRNA expression levels. Analyzing both layers of information provides insights into post-transcriptional regulation and functional relationships between genes and proteins.
- Comprehensive Analysis of Regulatory Mechanisms: Understanding the correlation between proteomic and transcriptomic data aids in deciphering regulatory mechanisms. Differential expression at the protein level, despite stable mRNA levels, suggests post-transcriptional regulation mechanisms such as microRNA-mediated control.
B. Multi-Omics Integration Platforms:
- Tools for Integrating Proteomics with Genomics, Transcriptomics, and Metabolomics: Platforms like OmicsIntegrator and IntegrOmics enable the integration of proteomics data with other omics layers. These tools facilitate a holistic systems biology approach, providing a comprehensive view of cellular regulation and interactions.
- Holistic Understanding of Cellular Regulation: Multi-omics integration platforms contribute to a holistic understanding of cellular processes by considering the interconnectedness of various molecular layers. This integrated approach enhances the identification of key regulatory nodes and their roles in cellular homeostasis.
IX. Machine Learning Approaches
- Machine Learning Algorithms for Predicting Protein Regulation: Utilizing machine learning algorithms, such as random forests, support vector machines, or neural networks, enables the construction of predictive models for protein regulation. These models leverage features derived from proteomics data to predict factors influencing protein abundance or modification.
- Feature Selection and Model Evaluation: Feature selection techniques, like recursive feature elimination or feature importance analysis, help identify the most relevant variables influencing protein regulation. Model evaluation metrics, including accuracy, precision, and recall, assess the performance of predictive models, ensuring their robustness and reliability in capturing regulatory patterns.
X. Challenges in Proteomics Data Analysis
A. Data Complexity:
- Handling Large-Scale, High-Dimensional Datasets: The vast amount of data generated in proteomics studies, especially with advancements like high-throughput mass spectrometry, poses challenges in storage, processing, and analysis. Implementing scalable solutions becomes crucial to manage and extract meaningful insights from large-scale proteomics datasets.
- Computational Challenges in Analysis: The intricate nature of proteomics data, involving complex relationships between proteins and their modifications, demands sophisticated computational tools. Addressing computational challenges requires the development and optimization of algorithms capable of handling the intricacies of proteomics data analysis efficiently.
XI. Future Trends and Innovations
A. Advancements in Mass Spectrometry:
- Improvements in Sensitivity and Resolution: Ongoing advancements in mass spectrometry technologies are expected to enhance sensitivity and resolution. Increased sensitivity allows the detection of low-abundance proteins, providing a more comprehensive view of the proteome. Improved resolution contributes to the accurate identification and characterization of closely related protein variants.
- Impact on Proteomics Data Analysis: These advancements will have a profound impact on proteomics data analysis by enabling the identification of a broader range of proteins and their modifications. Analytical techniques with higher sensitivity and resolution contribute to more accurate quantification and characterization of proteins, paving the way for deeper insights into cellular processes and regulatory mechanisms. Integrating these technological innovations into proteomics workflows will shape the future of data analysis in the field.
XII. Conclusion
A. Recap of Proteomics Data Analysis: In conclusion, the field of proteomics data analysis plays a crucial role in unraveling the complexities of cellular protein regulation. From the preprocessing of raw data to advanced differential expression analysis, pathway enrichment studies, and integration with other omics data, researchers are equipped with powerful tools to decipher the intricacies of the proteome.
B. Insights into Cellular Protein Regulation: The insights gained from proteomics data analysis extend beyond the identification and quantification of proteins. They provide a nuanced understanding of post-translational modifications, protein-protein interactions, and their integration with other cellular processes. As technology continues to advance, proteomics data analysis will remain at the forefront of scientific discoveries, contributing to our knowledge of cellular function and regulation.
Related posts:
![proteomics]()
Latest Breakthroughs in Targeted Protein Degradation
proteomics![bioinformatics-DNA, protein]()
Submitting High-Throughput Sequence Data to GEO (Gene Expression Omnibus)
bioinformatics![proteomics-omics]()
How Proteomics is Contributing to Vaccine Development
proteomics![proteomics-omics]()
Proteins: The Tiny Machines of Life - Demystifying Proteomics and its Role in Health and Disease
proteomics![Deepmind-Aplhafold]()
Protein Structure Determination and Prediction
proteomics![proteomics-quantative]()
Quantitative Proteomics Techniques
proteomics![Gene analysis tools and software]()
The Power of Protein Databases: A Comprehensive Guide to Their Vital Role in Proteomics
Guides![Proteomics tools]()
Introduction to Proteomics tools
proteomics![Artificial_Intelligence__AI__Machine_Learning_-_Deeplearning]()
How Deep Learning is Revolutionizing Omics?
bioinformatics![genomicsandproteomics]()
Proteomics and Genomics
genomics![3Dstructureofprotein-deepmind]()
Deep Learning in Drug Discovery: Predicting Protein-Ligand Binding Affinity
A.I![bioinformatics-DNA, protein]()
Genomics vs. Proteomics: Understanding the Key Differences
genomics![Strategies, Challenges, and Solutions in Multi-Omics Data Integration]()
Strategies, Challenges, and Solutions in Multi-Omics Data Integration
bioinformatics![Deepmind-Aplhafold]()
AI Revolution in Protein Structure Prediction
A.I![singlecelltranscriptomics]()
Advances in Transcriptomics and Proteomics
proteomics![AI-drug discovery]()
Proteomics in Drug Discovery: A Revolution in Pharma
proteomics