

How to Perform Protein Sequence Analysis?
December 3, 2024How to Perform Protein Sequence Analysis
Protein sequence analysis is a fundamental aspect of bioinformatics and molecular biology, enabling researchers to understand the structure, function, and evolutionary relationships of proteins. By analyzing the sequence of amino acids in a protein, scientists can predict its biochemical properties, identify conserved motifs, and uncover its role in biological processes. This analysis serves as a crucial step in many applications, including drug discovery, genetic research, and disease diagnosis.
Advances in computational tools and algorithms have streamlined the process, making it accessible to both experts and beginners in the field. This guide will explore the key steps and methods involved in protein sequence analysis, from retrieving sequences in public databases to performing alignment, structural predictions, and functional annotations. Understanding these techniques is essential for unlocking the insights hidden within protein sequences and translating them into meaningful biological knowledge.
The 20 Amino Acids and Their Role in Protein Structures
Overview
Each of the 20 most common amino acids has specific chemical characteristics and a unique role in protein structure and function. Based on the propensity of the side chains to be in contact with water (polar environment), amino acids can be classified into three groups:
1. Those with polar side chains.
2. Those with hydrophobic side chains.
3. Those with charged side chains.
Below we look at each of these classes and briefly discuss their role in protein structure and function.
Polar amino acids
When considering polarity, some amino acids are straightforward to define as polar, while in other cases, we may encounter disagreements. For example, serine (Ser, S), threonine (Thr, T), and tyrosine (Tyr, Y) are polar since they carry a hydroxylic (-OH) group. Furthermore, this group can form a hydrogen bond with another polar group by donating or accepting a proton (a table showing donors and acceptors in polar and charged amino acid side chains can be found at the FoldIt site.
Tyrosine is also involved in metal binding in many enzymatic sites. Asparagine (Asn, N) and glutamine (Gln, Q) also belong to this group and may donate or accept a hydrogen bond. Histidine (His, H), on the other hand, depending on the environment and pH, can be polar or carry a charge. It has two –NH groups with a pKa value of around 6. When both groups are protonated, the side chain has a charge of +1. Within protein molecules, the pKa may be modulated by the environment so that the side chain may give away a proton and become neutral or accept a proton, becoming charged. This ability makes histidine useful in enzyme active sites when the chemical reaction requires proton extraction. The aromatic amino acids tryptophan (Trp, W) and Tyr and the non-aromatic methionine (Met, M) are sometimes called amphipathic due to their ability to have both polar and nonpolar character. In protein molecules, these residues are often found close to the interface between a protein and solvent. We should also note here that the side chains of histidine, and tyrosine, together with the hydrophobic phenylalanine and tryptophan, can also form weak hydrogen bonds of the types OH−π and CH−O, using electron clouds within their ring structures. For a discussion of OH−π and CH−O kinds of hydrogen bonds, see Scheiner et al., 2002. A characteristic feature of aromatic residues is that they are often found within the core of a protein structure, with their side chains packed against each other. They are also highly conserved within protein families, with Trp having the highest conservation rate.
Hydrophobic amino acids
The hydrophobic amino acids include alanine (Ala, A), valine (Val, V), leucine (Leu, L), isoleucine (Ile, I), proline (Pro, P), phenylalanine (Phe, F) and cysteine (Cys, C). These residues typically form the hydrophobic core of proteins, which is isolated from the polar solvent. The side chains within the core are tightly packed and participate in van der Waals interactions, which are essential for stabilizing the structure. In addition, Cys residues are involved in three-dimensional structure stabilization through the formation of disulfide (S-S) bridges, which sometimes connect different secondary structure elements or different subunits in a complex. Another essential function of Cys is metal binding, sometimes in enzyme active sites and sometimes in structure-stabilizing metal centers. Also, in the case of Cys, some disagreement exists on its assignment to the hydrophobic group. For example, according to some schemes, it is found in the interior due to its high reactivity (Fiser et al., 1996), while others consider it polar (REF). The image below showing the distribution of buried/exposed fractions of amino acids in protein molecules suggests that Cys is rather exposed.
Charged amino acids
The charged amino acids at neutral pH (around 7.4) carry a single charge in the side chain. There are four of them; the two basic ones include lysine (Lys, K) and arginine (Arg, R), with a positive charge at neutral pH. The two acidic residues include aspartate (Asp, D) and glutamate (Glu, E), which carry a negative charge at neutral pH. A so-called salt bridge is often formed by the interaction of closely located positively and negatively charged side chains. Such bridges are often involved in stabilizing three-dimensional protein structure, especially in proteins from thermophilic organisms (organisms that live at elevated temperatures, up to 80-90 C, or even higher, REF). The binding of positively charged metal ions is another function of the negatively charged carboxylic groups of Asp and Glu. Metalloproteins and the role of metal centers in protein function is a fascinating field of structural biology research.
Glycine & proline
Glycine (Gly), one of the common amino acids, does not have a side chain and is often found at the surface of proteins within loop or coil regions (regions without defined secondary structure), providing high flexibility to the polypeptide chain. This flexibility is required in sharp polypeptide turns in loop structures. Proline, although considered hydrophobic, is also found at the surface, presumably due to its presence in turn and loop regions. In contrast to Gly, which provides the polypeptide chain high flexibility, Pro provides rigidity by imposing certain torsion angles on the segment of the structure (Morgan & Rubenstein, 2013). The reason for this is that its side chain makes a covalent bond with the main chain, which constrains the phi-angle of the polypeptide in this location (see the section of the Ramachandran plot). Sometimes Pro is called a helix breaker since it is often found at the end of α-helices. The importance of Gly and Pro in protein folding has been discussed by (Krieger et al., 2005).
The three-letter and one-letter codes for common amino acids
Charged amino acids (side chains often form salt bridges, in RED mnemonic rules to remember the one-letter code):
• Arginine – Arg – R
• Lysine – Lys – K
• Aspartic acid – Asp – D (AsparDic acid – hard t sound like D)
• Glutamic acid – Glu – E (GluEtamic acid – feels like it is almost an E there)
Polar amino acids (form hydrogen bonds as proton donors or acceptors):
• Glutamine – Gln – Q (Qlutamine – soft G)
• Asparagine – Asn – N (AsparagiNe – clear N)
• Histidine – His – H
• Serine – Ser – S
• Threonine – Thr – T
• Tyrosine – Tyr – Y
• Cysteine – Cys – C
Amphipathic amino acids (often found at the surface of proteins or lipid membranes, sometimes also classified as polar):
• Tryptophan – Trp – W (the largest side chain and the largest letter)
• Tyrosine – Tyr – Y
• Methionine – Met – M (may function as a ligand to metal ions)
Hydrophobic amino acids (normally buried inside the protein core):
• Alanine – Ala – A
• Isoleucine – Ile – I
• Leucine – Leu – L
• Methionine – Met – M
• Phenylalanine – Phe – F
• Valine – Val – V
• Proline – Pro – P
• Glycine – Gly – G
The distribution of amino acids in proteins
The preferred location of different amino acids in protein molecules can be quantitatively characterized by calculating the extent to which an amino acid is buried in the structure or exposed to solvent. The image below provides an overview of the distribution of the different amino acids within protein molecules. While hydrophobic amino acids are mostly buried, a smaller number of polar side chains are buried, while charged residues are mostly exposed.
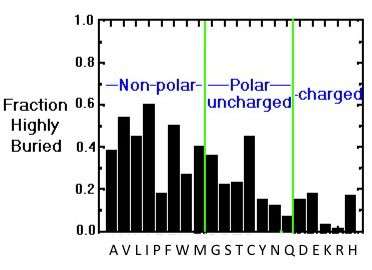
Buried/exposed fraction of amino acids within protein molecules.
The vertical axis shows the fraction of highly buried residues, while the horizontal axis shows the amino acid names in one-letter code. Image from the tutorial by J.E. Wampler,
A discussion on the relationship between surface accessibility and the conservation of amino acids can be found in Fiser et al, 1996.
Water & hydrogen bonds
A few words about hydrogen bonds: for a hydrogen bond to be formed, two electronegative atoms (for example, in the case of an α-helix, the amide nitrogen, and the carbonyl oxygen) must interact with the same hydrogen. The hydrogen is covalently attached to one of the atoms (called the hydrogen bond donor) and interacts with the other (the hydrogen bond acceptor). In proteins, all groups capable of forming H-bonds (both main chain and side chain) are usually H-bonded to each other or to water molecules (REF). Due to their electronic structure, water molecules may accept two hydrogen bonds and donate 2, thus being simultaneously engaged in a total of 4 hydrogen bonds. Water molecules may also stabilize protein structures by making hydrogen bonds with the main chain or side chain groups in proteins and even mediating the interactions of different protein groups by linking them to each other by hydrogen bonds. In addition, water is often involved in ligand binding to proteins, mediating ligand interactions with polar or charged side chains or main chain atoms. It is helpful to remember that the energy of a hydrogen bond depends on the distance between the donor and the acceptor and the angle between them and is in the range of 2-10 kcal/mol. The work by Hubbard & Haider, 2010 can be recommended for more detailed information on hydrogen bonds.
What is Protein Sequence?
Proteins are fundamental macromolecules that serve as the workhorses of the cell, participating in virtually every biological process. A protein sequence is a specific order of amino acids linked by peptide bonds, forming the backbone of the protein structure. The unique order of amino acids in a protein sequence is crucial because it dictates how the polypeptide chain will fold into its specific three-dimensional structure. This structure is essential for the protein’s function, as the spatial arrangement of amino acids within the protein determines how it interacts with other molecules and carries out its biological roles. Any alterations in the protein sequence, such as mutations, can lead to changes in structure and function, potentially resulting in various diseases or altered biological activities.
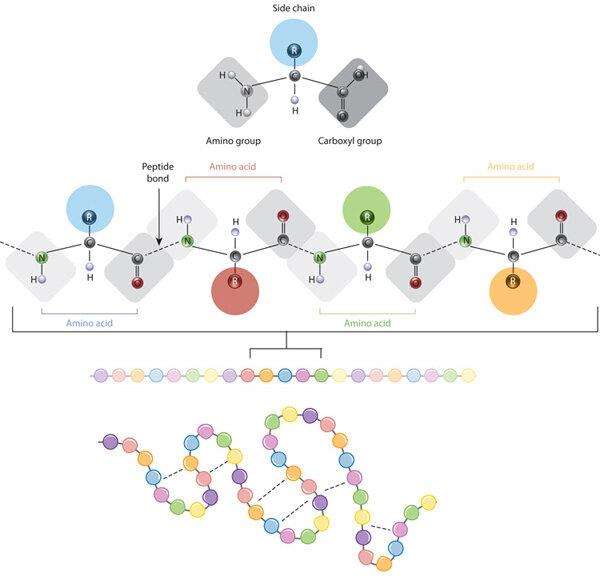 The relationship between amino acid side chains and protein conformation (Giancarlo Croce, 2019)
The relationship between amino acid side chains and protein conformation (Giancarlo Croce, 2019)
The Four Levels of Protein Structure
Primary Structure of Proteins
The primary structure of a protein is its most basic and fundamental level, comprising a specific and linear sequence of amino acids in a polypeptide chain. This sequence is determined by the nucleotide sequence of the gene encoding the protein. The primary structure is critically important because it dictates the higher levels of protein structure—secondary, tertiary, and quaternary—and ultimately the protein’s function. Each amino acid in the sequence has distinct chemical properties, influencing how the chain will fold and interact with itself and other molecules.
Secondary Structure of Proteins
The secondary structure of a protein refers to the local folding patterns of the polypeptide chain, which occur due to interactions between the backbone atoms of the amino acids. The most common secondary structures are alpha helices and beta sheets. Alpha helix is compact and rigid, often found in regions of proteins that require stability, such as membrane-spanning domains. Beta sheets are typically found in the core of globular proteins and in fibrous proteins like silk. In addition to alpha helices and beta sheets, other secondary structures include turns and loops, which connect the helices and sheets, allowing the polypeptide chain to fold into a compact structure.
Tertiary Structure of Proteins
The tertiary structure represents the three-dimensional conformation of a single polypeptide chain, formed by the overall folding and interactions of the secondary structural elements. This level of structure is stabilized by various interactions, including hydrophobic interactions, hydrogen bonds, and disulfide bridges. The tertiary structure is critical because it determines the protein’s specific function. The precise folding of the polypeptide chain creates the unique three-dimensional pocket that can accommodate the substrate with high specificity. Any alteration in the tertiary structure, whether due to mutation or environmental changes, can disrupt the protein’s function, leading to diseases or loss of biological activity.
Quaternary Structure of Proteins
The quaternary structure pertains to the assembly of multiple polypeptide chains (subunits) into a functional protein complex. This level of structure is essential for the activity of multimeric proteins, such as hemoglobin. The formation of a quaternary structure allows for cooperative interactions between subunits, which can enhance the protein’s functionality. Moreover, quaternary structures enable the regulation of protein activity through allosteric effects, where the binding of a molecule at one site on the protein affects the activity at another site. This is particularly important in enzymes and signaling proteins, where such regulation allows the cell to respond to changes in its environment.
Methods of Protein Sequencing
Protein sequencing aims to determine the amino acid sequence of proteins. The methods used for protein sequencing can be broadly categorized into traditional techniques, advanced techniques, and specific approaches for single-protein sequencing.
Classical Protein Sequencing Methods
Edman Based Protein Sequencing
Edman degradation is a chemical process that sequentially removes one amino acid residue at a time from the N-terminus of a peptide or protein. This method uses phenylisothiocyanate (PITC) to label the N-terminal residue, which is then cleaved and identified by chromatography.
Mass Spectrometry Based Protein Sequencing
Mass Spectrometry (MS): Mass spectrometry determines the mass-to-charge ratio of peptides and proteins, providing information about their molecular weight and sequence. Techniques include:
Matrix-Assisted Laser Desorption/Ionization (MALDI): A technique where peptides are ionized and analyzed in a mass spectrometer after being mixed with a matrix material that absorbs laser energy.
Electrospray Ionization (ESI): A technique that uses an electric field to generate ions from peptides in solution.
Tandem Mass Spectrometry (MS/MS): Tandem mass spectrometry involves multiple stages of mass spectrometry to fragment peptides into smaller ions. This allows for detailed sequencing of peptides by analyzing the fragment ions.
De Novo Sequencing: De novo sequencing involves determining peptide sequences without relying on prior sequence information. This method utilizes mass spectrometry to analyze the fragmentation patterns of peptides.
Integrated and Emerging Methods
Edman Degradation Combined with MS: This approach combines the strengths of Edman degradation and mass spectrometry to provide enhanced sequencing capabilities. Edman degradation is used for initial sequence determination, while mass spectrometry helps confirm and extend the sequence
Single-Molecule Protein Sequencing: Utilizes nanopore sequencing technologies to analyze individual protein molecules in real-time. Proteins are passed through a nanopore, and their sequence is inferred based on disruptions in ionic current.
Comparing Protein Sequencing Methods
| Method | Advantages | Limitations |
| Edman Degradation |
|
|
| Mass Spectrometry (MS) |
|
|
| Tandem Mass Spectrometry (MS/MS) |
|
|
| De Novo Sequencing |
|
|
| Edman Degradation Combined with MS |
|
|
| Single-Protein Sequencing |
|
|
| Single-Molecule Protein Sequencing |
|
|
| Single-Cell Proteomics |
|
|
- Protein Sequencing: Methods, Applications, and Implications
Techniques for Analyzing Protein Sequences
Bioinformatics Tools and Databases for Protein Analysis
BLAST: BLAST is a widely used algorithm for comparing a protein sequence against a database of known sequences. By identifying homologous sequences, BLAST provides insights into the potential function of the query protein based on known functions of similar proteins. It can be used to identify evolutionary relationships and predict functional domains.
UniProt: UniProt is a comprehensive protein sequence database offering detailed annotations for proteins from various organisms. It includes information on protein function, structure, post-translational modifications, and interactions. UniProt is crucial for functional annotation, as it provides context and background information for proteins of interest.
Pfam and InterPro: Pfam is a database of protein families, each defined by a conserved sequence motif or domain. InterPro integrates multiple protein family and domain databases, including Pfam, to provide a unified view of protein domain architectures. These databases are essential for understanding the functional roles of protein domains and predicting protein functions based on domain composition.
Sequence Alignment and Comparison
Clustal Omega: Clustal Omega is a powerful tool for multiple sequence alignment, which aligns three or more protein sequences to identify conserved regions and infer evolutionary relationships. By comparing sequences across different species or within protein families, Clustal Omega helps in identifying conserved motifs that are crucial for protein function.
MUSCLE: MUSCLE (Multiple Sequence Comparison by Log-Expectation) is another widely used tool for multiple sequence alignment. It offers high accuracy and efficiency in aligning large sets of sequences, making it suitable for detailed comparative analyses and functional predictions.
Functional Annotation of Protein Sequences
InterProScan: InterProScan is a tool that integrates various protein signature databases, such as Pfam, PRINTS, and PROSITE, to predict the functional annotations of protein sequences. It provides comprehensive information on functional domains, family memberships, and predicted biological roles, facilitating deeper understanding of protein functions.
Gene Ontology (GO): Gene Ontology provides a structured vocabulary to describe the functions of proteins in terms of biological processes, cellular components, and molecular functions. By mapping protein sequences to GO terms, researchers can gain insights into the biological roles of proteins and their involvement in cellular pathways.
Protein Sequence Variation and Polymorphisms
Understanding Protein Sequence Variations
Protein sequence variations arise from changes in the DNA that encodes for the protein. The most common types of variations include:
Single nucleotide polymorphisms (SNPs): SNPs are the most frequent type of genetic variation, involving a single base pair change in the DNA sequence.
Insertions and deletions (Indels): Indels are the insertion or deletion of one or more nucleotides in the DNA sequence.
Structural Variants: These are larger changes in the DNA, such as duplications, inversions, or translocations of entire segments of chromosomes.
Protein sequence variations can arise through various mechanisms:
Mutations: Spontaneous errors during DNA replication can introduce mutations, leading to changes in the protein sequence.
Genetic Recombination: During meiosis, homologous chromosomes exchange segments of DNA in a process known as recombination.
Gene Duplication: Sometimes, entire genes are duplicated, leading to additional copies of a protein-coding gene.
Impact of Variations on Protein Function
Protein sequence variations can have diverse effects on protein structure and function, depending on the nature and location of the change:
Altered amino acid properties: A single amino acid change can significantly impact protein folding, stability, or interactions with other molecules.
Disruption of active sites: Variations in critical regions of the protein, such as active sites or binding sites, can impair the protein’s ability to catalyze reactions or interact with other molecules, leading to loss of function.
Gain of new functions: In some cases, sequence variations can result in proteins with new or enhanced functions.
The Importance of Protein Sequences
How Protein Sequences Determine Function
The composition of amino acids in a protein determines its unique three-dimensional structure, which in turn dictates its specific biological function. Proteins fold into complex structures driven by interactions between amino acid side chains, including hydrogen bonds, hydrophobic interactions, and ionic bonds. Alterations in the amino acid sequence, even single-point mutations, can significantly affect a protein’s folding, stability, and functional capabilities. For instance, signal peptides are short amino acid sequences located at the beginning of nascent proteins. They direct the protein to its correct cellular destination, such as the endoplasmic reticulum for secretion. Alterations in the signal peptide sequence can lead to mislocalization or malfunction of the protein, impacting its role in glucose regulation.
- Single Molecule Protein Sequencing
Protein Sequences and Disease Mechanisms
Mutations or aberrations in protein sequences can result in proteins that are misfolded or functionally compromised, leading to a variety of diseases. For example, in sickle cell anemia, a mutation in the hemoglobin protein, where the amino acid glutamic acid is replaced by valine at the sixth position of the beta-globin chain, leads to the abnormal folding of the hemoglobin molecule. This misfolding causes the red blood cells to assume a sickle shape, impairing their ability to carry oxygen and leading to severe health complications. By analyzing protein sequences, researchers can identify disease-causing mutations, understand their impacts on protein function, and develop targeted diagnostic tools and treatments. This approach is crucial for advancing personalized medicine and developing therapies tailored to specific genetic alterations.
Applications of Protein Sequences in Drug Design
Understanding protein sequences is pivotal in drug design, particularly for targeted therapies. By analyzing protein sequences, drugs can be tailored to interact precisely with specific proteins, enabling precision medicine approaches.
One of the most successful applications of protein sequence in drug design is the development of kinase inhibitors for cancer therapy. Kinases are enzymes that play a crucial role in cell signaling, and their dysregulation is often associated with cancer. By analyzing the amino acid sequences of specific kinases, researchers can identify unique features of their active sites that can be targeted by drugs. Furthermore, biopharmaceutical variation analysis is essential for identifying sequence variations that may impact drug efficacy and safety. This analysis ensures that drugs are optimized for individual patients, enhancing effectiveness, reducing off-target effects, and improving patient outcomes.
Evolutionary Biology and Phylogenetics
The comparison of protein sequences across different species is a powerful tool in evolutionary biology, providing insights into the conservation and divergence of protein functions over time. By analyzing protein sequences, scientists can trace the evolutionary history of proteins, identify conserved functional domains, and understand the selective pressures that have shaped protein evolution.
Phylogenetic analysis of protein sequences can help identify key evolutionary events, such as gene duplications, that have led to the diversification of protein families. For example, the globin family of proteins, which includes hemoglobin and myoglobin, has undergone several gene duplication events, leading to the evolution of proteins with specialized functions in oxygen transport and storage. By studying the sequences of globin proteins in different species, researchers can infer the evolutionary history of these proteins and understand how they have adapted to the specific needs of different organisms.
Introduction to Protein Sequence Alignment and Analysis
Theoretically, retrieving most of the information we need about a protein directly from its amino acid sequence should be possible. To do this, we need sequence analysis. However, the study of sequence alignment can be challenging and requires prior knowledge and experience. Sometimes, we can compare such study to attempting to read a text in a foreign language without speaking it: we may recognize the letters and even some words, but we are not going to understand the meaning of the text. Since our focus here is on structural biology and the relationships between the amino acid sequence, structure, and function, you will only find an overview of the basic concepts and techniques used in sequence alignment and analysis without going into the theory and algorithms behind the process. We primarily use sequence alignment to extract structural and functional information from a sequence and understand the proteins’ evolutionary relationships.
Before running sequence alignment and analysis, we need to understand the essential characteristics of the 20 most common amino acids that build up the proteins we find in nature. The amino acids can be classified into three main groups: hydrophobic, polar, and charged. Different amino acids are responsible for different functions in proteins. Some are often found at functional sites; hydrophobic amino acids usually build up the core of protein molecules, while charged residues are distributed on the protein surface in contact with a solvent. A sequence alignment will quickly reveal the distribution of these residues along the protein amino acid sequence and will also show their conservation pattern in a protein family.
Many positions in a protein sequence change during evolution due to mutations in the genetic code. However, many of these substitutions are so-called conserved substitutions. This means that other hydrophobic amino acids usually replace hydrophobic amino acids, which also applies to charged and polar amino acids. In other words, we observe the conservation of the chemical nature of an amino acid. This raises the question of how we account for these substitutions when running and scoring a sequence alignment. Mutation probability matrices (substitution matrices) were introduced to answer this question. These matrices consider the frequency of amino acid substitution replacements in calculating an alignment score.
The first step in sequence alignment and analysis is placing the protein we are interested in within a specific frame, which is the protein family to which it belongs. Within a family, proteins perform a similar function, have conserved sequence features, conserved three-dimensional structures, etc. An alignment will help us reveal all these characteristic features, find the conserved and variable regions in the sequence, show functionally essential residues, extract information on the secondary and tertiary structure, etc. We will discuss the techniques used for performing sequence alignment and analysis and demonstrate the process’s practical aspects in a tutorial.
For an alignment, we first need to find related sequences. To do this, we need to run a database search. The software will compare our sequence (query sequence) to all other sequences in the database by using a local alignment algorithm BLAST and, based on specific criteria, will output several amino acid sequences found to be related to our protein. Local alignment compares small segments of the query sequence to other sequences in the database to find matching amino acid segments. If we are happy with the search results and have a list of proteins to start the analysis with, we can run a so-called global alignment. In this case, we can run a pair-wise alignment of only two sequences or a multiple sequence alignment (MSA) using a group of sequences. MSA constitutes the basis of any sequence analysis and provides much more information than the alignment of a pair of sequences. It is extensively used in secondary and tertiary structure prediction and modeling to identify protein families and domains.
Local alignment compares small segments of the query sequence to other sequences in the database to find matching amino acid segments. I usually use the Expasy and EBI servers, although many others perform the same function. The image below shows an example of a BLAST-generated local alignment (66 residues) made on the UniProt server.

An example of a local sequence alignment (residues 1 to 66 included) was generated by a Uniprot search using the sequence of the magnesium chelatase BchI subunit.
Amino Acid Substitutions & Replacement Matrices
Mutation prabability matrix
The previous page discussed the need for a scoring scheme to score amino acid replacements for an accurate sequence alignment. A scoring scheme only based on identities is not very effective. It will miss much helpful information, especially when we need to detect weak homology. When discussing amino acid replacement schemes, we must remember that during evolution, amino acids may be replaced by the original type after several cycles. Such a process is logical since the conservation of structure and function always has higher priority in the selection process. This type of conservation has also been described as conservation of size and hydrophobicity (Taylor, 1986).
Margaret Dayhoff and co-workers, who pioneered the field of protein sequence analysis, databases, and bioinformatics, developed the first mutation probability matrix in the 1970s. Their model was based on observed frequencies of substitutions of each of the 20 amino acids derived from an alignment of closely related sequences. Based on the number of common amino acids (20), we can count 210 possible replacement pairs, of which 190 pairs correspond to two different amino acids and 20 pairs of identical substitutions. In the resulting so-called mutation probability matrix, each element Mij provides an estimate of the probability of an amino acid in column j to be mutated to the amino acid in row i after a particular evolutionary time.

Image from Byron Holland: https://slideplayer.com/slide/8888934/
An evolutionary unit of 100 million years was adapted, resulting in the PAM (Percentage Accepted Mutations / 100 million years) matrix. 1 PAM corresponds to an average amino acid substitution in 1% of all positions. Although 100 PAM does not mean that all the amino acids in the sequence are different (as compared to the original sequence), since, as noted above, many of them will be mutated back to their original type.
Further analysis showed that, although at 256 PAM, 80 % of all amino acids will be substituted, 48% of Trp, 41% of Cys, and 20% of His will be conserved. On the other hand, only 7% of S residues will remain (discussed in Barton, J, 1996). This conservation pattern presumably results from a combination of structural and functional restraints mentioned above. For example, tryptophan has a large side chain, and if positioned within the structure’s core, its replacement by another amino acid may destabilize the protein. In addition, the other highly conserved residues, cysteine and histidine, are often involved in specific functions like proton abstraction (His), metal binding (His and Cys), or disulfide bridge formation (Cys).
After the construction of the mutation probability matrix, Dayhoff et al. defined the score Si,j of two aligned residues i, j according to the following equation:

In which (Mij ) is the probability of these two residues being aligned (from the matrix above), and pi – the probability of these two amino acids being aligned by chance.
This type of scoring will give higher numbers to the alignment of a pair of similar amino acids (e.g., Leu and Ile) and lower numbers when the amino acids are different (like I and D). Using this definition, the probability matrix (shown in the image above) can then be used to derive the so-called log-odds scoring matrix:

This replacement matrix can be used for the calculation of the alignment score according to the equation discussed earlier: (score) S= Σ of costs (identities, replacements) – Σ of penalties (number of gaps x gap penalties)
PAM, PET91 & BLOSUM matrices
Different amino acid substitution matrix versions have been generated and serve different purposes. For example, low PAM matrices (20, 40, 60) are preferred in database scanning when short, closely related sequence stretches need to be found. On the other hand, higher PAM matrices, are more suitable for aligning longer sequences. Many internet resources for sequence alignment provide the option of choosing the substitution matrix for the alignment.
The original Dayhoff matrix was based on a limited set of known protein sequences, and no statistical data could be collected for many of the possible 190 substitutions. This was corrected later when more data became available. An example is PET91, an updated Dayhoff matrix (Jones et al., 1992) based on 2,621 protein families from the SwissProt database (now UniProt, part of the Expasy server). Meanwhile, other substitution matrices were developed based on slightly different principles. One of the most popular is the BLOSUM matrix (BLOcks of Amino Acid SUbstitution Matrix, Henikoff S, Henikoff JG. 1992). BLOSUM scores amino acid replacements based on the frequencies of amino acid substitutions in ungapped aligned blocks of sequences with a specific percentage identity. Thus, there is a significant difference between PAM and BLOSUM matrices – PAM matrices are based on mutations observed in a global alignment, which includes highly conserved regions and low-conservation regions with gapped alignment. The numbers associated with each matrix (e.g., BLOSUM62, BLOSUM80, etc.) refer to the minimum percentage sequence identity for the sequences aligned within a block. Higher numbers correspond to higher sequence identity and shorter evolutionary distances between the proteins. BLOSUM with high numbers should then be used for sequences known to be homologous with a good level of sequence identity. At the same time, low BLOSUM numbers should be used for distantly related proteins, for example, in screening databases.
Structure-based alignment
The 3D structure of a protein contains vital information on the position and length of secondary structure elements and loop regions. Since many gaps in sequence alignment occur in regions between secondary structure elements, using information from a 3D structure will provide a more accurate positioning of insertions and deletions in the alignment. Many graphics programs include the superposition of 3D structures and provide a structure-based alignment of the sequences as an output. However, this alignment still needs to be verified, e.g., by examining the 3D structure and comparing the generated alignment to an automatic multiple sequence alignment.
Concluding remarks
This brief review explains some of the basic concepts in the sequence alignment. Since, as mentioned earlier, here we are focused on the practical application of the tools for sequence alignment and analysis, I did not get into the details of the statistical analysis behind the construction of the replacement matrices, the data related to sequence conservation, or details of calculating alignment scores. In a tutorial, we will discuss sequence alignment’s practical aspects.
Sequence Alignment and Analysis Tutorial
Background
Amino acid sequence alignment is a great tool in the study of proteins. Nowadays, we can find much information about a protein using only the sequence. With the arrival of AlphaFold and the ESM Metagenomic Atlas, even the predicted three-dimensional structure is now available for our analysis. Still, alignment of the amino acid sequence may provide valuable insights into the protein’s conservation pattern, the presence of functionally important conserved motifs, domain structure, evolutionary relationships within the family, etc.
This tutorial will analyze two related proteins, two subunits of the enzyme magnesium chelatase, BchI and BchD. I will use the tools available at UniProt and EBI multiple sequence alignment (MSA) servers. Although many other servers will do the same job, I am used to these two. We will explore how to make a sequence alignment. At the same time, we will examine the information in UniProt and other related databases to learn more about these two proteins.
Subunits BchI (35 kDa) and BchD (70 kDa) together build up a large 600 kDa complex ; there is also a third subunit required for the enzyme’s function called BchH (about 120 kDa). However, we are not going to discuss it here. Magnesium chelatase catalyzes the insertion of an Mg2+ ion into protoporphyrin IX at the first committed step of chlorophyll biosynthesis. As we can guess from the size, BchI, and BchD are multidomain proteins. In such cases, we need to define the domains and, by analyzing any conservation patterns, try to identify possible relationships between them and other proteins. Insights gathered from such analysis may help us better understand the mechanism of enzyme function. There are, of course, different ways of running such analysis. I will be showing just one which I think is logical. After learning how to use the tools described here, you will be able to design your analysis routines, whatever the aim of your analysis may be.
Finding a sequence: Running Blast
In the beginning, we need to choose the sequences for the alignment. To start, we write the name of the protein (BchD) into the UniProt search window, and we will be taken to a list of protein sequences from different organisms.
The site shows a large number of sequences. You may notice on the left, where it says “Status,” that there are “Reviewed” and “Unreviewed” sequences. This is one of my favorite features – when there is a sufficient number of reviewed sequences, I usually choose them for further analysis. These sequences have been verified to be what we expect them to be. Many automatically annotated sequences are among the Unreviewed, and sometimes we may find assignment errors. There is more helpful information (on the left field), like the availability of a 3D structure, catalytic activity, etc.
We will choose BCHD_RHOCB (entry P26175), the subunit BchD from Rhodobacter capsulatus. In plants, the homologous subunit is called ChlD; “B” in BchD indicates that the protein is involved in bacteriochlorophyll syntheses, while ChlD is related to chlorophyll synthesis.
On the page that opens after clicking the entry ID P26175, we will find detailed information on magnesium chelatase—its biological function (photosynthesis, magnesium chelatase activity), the type of ligands/substrate it binds, its catalytic function (insertion of magnesium and ATP hydrolysis), links to published works, links to entries related to this particular protein in other databases, and, of course, the amino acid sequence of BchD.
Clicking “Family & Domains” on the menu on the left of the UniProt page will show the domain content of BchD. However, in this case, we only get the vWFA domain (von Willebrand Factor A-like domain superfamily) at the C-terminal of the protein (residues 379-559). On the other hand, the InterPro database link directs us to the BCHD_RHOCB page, where we find a more detailed analysis of the domain composition of the protein. Apart from the vWFA domain, they also identify a P-loop NTPase family domain at the N-terminal part of the sequence, residues 78-235. The characteristic P-loop sequence motif, which is [AG]-x(4)-G-K-[ST] according to the Prosite database, is not conserved in R. capsulatus BchD; we will look closer into this when we analyze the sequence alignment. There is also a link to the AlfaFold predicted structure of the subunit, where we can explore the two domains and see their proposed arrangement in three dimensions.
A characteristic feature of the vWFA domain is the conservation of the so-called MIDAS motif. MIDAS stands for metal ion-dependent adhesion site (see a paper by Lacy et al. 2004 for a detailed structure description). The conserved sequence consists of the DXSXS motif and additional threonine and aspartate (T and D) residues. The DXSXS motif residues in BchD are D385, S387 S389, which are close to the N-terminus of the vWFA domain, while threonine T452 and aspartate D482 are located further down in the sequence. The alignment in the image below (click to see it, it can also be found at the Conserved Protein Domain Family server at NCBI) shows the position of these invariant residues (yellow boxes, also marked by a # on top of the alignment). These residues are involved in metal ion binding, Ca2+ or Mg2+. We still do not know the exact function of this domain within the magnesium chelatase complex.
Click for image
Moving closer to the bottom of the UniProt page we will find the sequence of the protein:

Since we need several sequences for the multiple sequence alignment, I usually run BLAST (Basic Local Alignment Search Tool) to get a list of related sequences. To run Blast, we can choose “Tools” (shown in the image above in the left corner) and there click “Blast.” On the Blast page, you may also choose the substitution matrix you want to use in the search. Blast searches the entire database and outputs a list of sequences aligned against our query sequence (pairwise alignment). I choose several sequences from the list according to organism and percentage sequence identity and save them in the Basket.
Click below to see part of the list after running Blast. In the image, we can see the sequence identity with the individual organism, and clicking there will show the alignment of the query sequence to the protein found in the database (figure below).
Click for image

Making the alignment
For the alignment, a good strategy is to choose 3-4 sequences since this will give a better idea of the conservation pattern in the protein family. To run the alignment, I mark the sequences I saved in the Basket and choose “Align” from the Basket menu. We will be taken to the alignment page. A helpful option is related to the order of sequences in the alignment output (Output sequence order) under “Advance parameters.” I prefer to use the “input order” to group bacterial and plant sequences separately. This will simplify the analysis of the conservation pattern. We can also edit the sequences in the alignment window if we need.
In the alignment window, the sequences are in the FASTA format, which is different from the format shown as default on the UniProt page. If we need the sequence in FASTA format, we need to click “Download” above the sequence shown in UniProt, then save it by copying and pasting it for later use or insert it into the MSA server alignment window if we choose to run the alignment there. At the bottom of this page, you will find an explanation of the FASTA format.
I usually remove most of the information on top of the sequence (everything which comes after the “larger than” sign > and only leave the name of the protein, e.g., BCHD_RHOCB. This will simplify the analysis of the alignment. Don’t forget to include the protein from Rhodobacter capsulatus.
Click below to see the alignment which I made.
Click for image
Analyzing the alignment
The alignment is colored according to similarity. There are other options for coloring, e.g., by residue type, which will show hydrophobic, charged, and polar amino acids. We can see that the sequence of BchD is well-conserved among different bacteria – there are just a few insertions and deletions.
We can also examine the AlphaFold predicted structure of BchD (image below) to get additional insights into the sequence-structure relationships for this protein. As mentioned above, the C-terminal domain (residues 375-559) was earlier recognized as a vWAF-type domain. The predicted structure shows that a long stretch of the sequence that does not seem to have much secondary structure connects the C-terminal domain to the N-terminal domain. The residues from approximately 232 to 309 (the orange color means prediction is unreliable) are followed by a region with some secondary structure which continues until Met375, where the vWAF domain starts. Looking at the alignment, we can see that up to Glu309 we have a so-called low-complexity region – it is not well conserved, and there are many repeats but also many acidic residues. Such regions are usually very flexible and often found to be involved in protein-protein interactions. This suggests that this region may somehow interact with other subunits within the large 600 kDa magnesium chelatase complex. The rest of the sequence, up to the vWAF domain, appears to be more structures, although judging by the color, the reliability of the prediction is still low there. The N-terminal domain, which contains residues 1-232, as demonstrated in the above-mentioned publication by Fodje et al., has a conserved fold of the so-called AAA+ family of proteins to which the smallest magnesium chelatase subunit BchI belongs. It is possible to verify this by submitting the AlphaFold predicted structure to one of the fold recognition servers like DALI. We can also verify the homology between this domain by an alignment of the sequences of BchD and BchI.

For the comparison, I have made an alignment that included BchI sequences. The sequence similarity is about 29%, which is not very high. The alignment (click to see it, only part of the BchD sequence is shown) also shows that the ATP binding site residues (the P-loop motif, marked on the alignment) of BchI are not conserved in BchD. This means that BchD does not hydrolyze ATP. However, it is still possible that ATP may bind to the protein and trigger oligomerization of the complex (it does for BchI). The alignment also shows large insertions in the BchI sequence compared to BchD. This is another reason for submitting the predicted structure of BchD to a fold recognition server. This way, we can verify that we indeed have a similar fold.
Click for image
Final notes
This tutorial demonstrates what can be done using the sequence analysis tools at UniProt and other servers, and including a predicted AlphaFold structure. One of the problems of AlphaFold predictions is that the structures are not classified in the CATH database. Hopefully, at some moment, this will change. Until then, we need to rely on our own analysis for the identification of domain fold in the predicted structures.
I am sure that there are many other ways to combine sequence and structure analysis. In addition, our analysis depends on our aim and what we try to achieve. I hope that this introduction will help you in your search for information.
The FASTA format
As noted above, many applications require the amino acid sequence to be in FASTA format. The FASTA format includes the amino acid sequence in one-letter code, usually with 60 letters/line. Most important is the sign “>,” “larger than,” on the first line. Alignment programs at the EBI MSA server will use the text after the >-sign on that line as the alignment title for the sequence. For convenience, one could write the name of the protein on that row, which would be helpful as a sequence identifier after running the alignment. BchI sequence in FASTA format is shown below:
>sp|P26239|BCHI_RHOCB Magnesium-chelatase 38 kDa subunit OS=Rhodobacter capsulatus (strain ATCC BAA-309 / NBRC 16581 / SB1003) OX=272942 GN=bchI PE=1 SV=1
MTTAVARLQPSASGAKTRPVFPFSAIVGQEDMKLALLLTAVDPGIGGVLVFGDRGTGKST
AVRALAALLPEIEAVEGCPVSSPNVEMIPDWATVLSTNVIRKPTPVVDLPLGVSEDRVVG
ALDIERAISKGEKAFEPGLLARANRGYLYIDECNLLEDHIVDLLLDVAQSGENVVERDGL
SIRHPARFVLVGSGNPEEGDLRPQLLDRFGLSVEVLSPRDVETRVEVIRRRDTYDADPKA
FLEEWRPKDMDIRNQILEARERLPKVEAPNTALYDCAALCIALGSDGLRGELTLLRSARA
LAALEGATAVGRDHLKRVATMALSHRLRRDPLDEAGSTARVARTVEETLP
Related posts:
![blastquery]()
Basic Local Alignment Search Tool (BLAST) for bioinformatics
bioinformatics![ChatGPT in healthcare and scientific applications]()
AI in Healthcare and Scientific Writing
A.I![3Dstructureofprotein-deepmind]()
Advances in Computational Molecular Modeling
bioinformatics![SARS-COV-2 virus]()
Best Programs for Phylogenetic Tree Visualization of Large Datasets
bioinformatics![Earthbiogenomeproject_omicstutorials]()
Sequencing the Future: AI, the Earth Biome Project, and the New Frontiers of Genomic Exploration
A.I![remotecomputer-bioinformatics]()
How To Efficiently Parse A Huge Fastq File?
bioinformatics![AI based search]()
Introduction to Information Technology
bioinformatics![Blast bioinformatics]()
Blast bioinformatics
bioinformatics![Precision-Medicine-Featured]()
What does precision medicine aim to achieve and how does bioinformatics enable it?
bioinformatics![bioinformatics jobs]()
Climbing the Career Ladder in Bioinformatics: Skills and Tips for Getting Ahead
bioinformatics![bigdatainbiology-omicstutorials]()
Mining Massive Biological Datasets for New Discoveries with Scalable Algorithms
bioinformatics![bioinformatics software]()
Automated Bioinformatics Analysis: A Comprehensive Bash Script Pipeline for Protein Analysis
bioinformatics![will bioinformatics replace by AI]()
Revolutionizing Bioinformatics for Beginners: A Journey with ChatGPT
A.I![AI based search]()
Bioinformatics for Intermediate Students: Setting Goals, Learning to Code, and Exploring Advanced To...
bioinformatics![spatialcomputing-bioinformatics]()
Introduction to Spatial Computing and its Applications in Bioinformatics
bioinformatics![Combining two plots, a histogram and a scatter plot, with `par()` function.]()
Introduction to R for Genomic Data Analysis
bioinformatics
















