Sequence Alignment in Bioinformatics: Types & Methods
October 8, 2024Sequence alignment is a fundamental tool in bioinformatics, used to compare biological sequences such as DNA, RNA, or proteins. It involves arranging two or more sequences to identify regions of similarity, which can reveal important functional, structural, or evolutionary relationships.
In bioinformatics, sequence alignment is essential for comparing newly obtained sequences with those already known and stored in databases.
There are two main types of sequence alignment:
- Global Alignment: This method aligns two sequences across their entire length, maximizing the overall similarity. It is typically used when comparing sequences of the same length, such as in genome-wide comparisons.
- Local Alignment: Local alignment focuses on aligning only the regions of sequences that show the highest degree of similarity, rather than the entire sequence. This method is particularly useful for identifying conserved regions in protein or nucleotide sequences that may be functionally or evolutionarily significant.
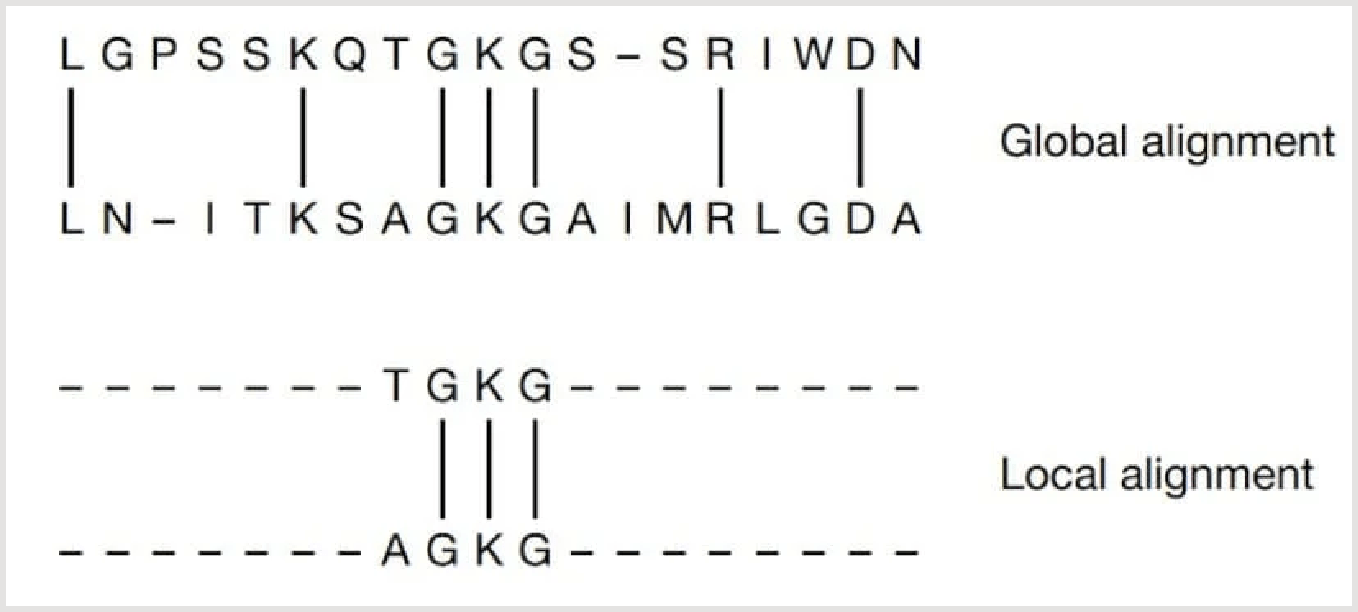
Figure: Global and Local Alignment of two sequences (Mount, D. W., 2001).
Types of Sequence Alignment
- Pairwise Alignment:
Pairwise sequence alignment involves aligning two sequences to identify the optimal pairing and similarity between them. It uses a scoring system where matching characters receive positive scores, while mismatches or gaps are penalized with negative scores. The main goal of pairwise alignment is to achieve the highest possible alignment score, reflecting the degree of similarity between the sequences. - Multiple Sequence Alignment (MSA):
Multiple sequence alignment extends this concept to three or more biological sequences. MSA is used to identify conserved regions across different sequences, which can provide insight into their functional, structural, or evolutionary relationships. It is also instrumental in constructing phylogenetic trees, which trace evolutionary connections among species or groups of organisms.
Methods of pairwise sequence alignment
There are three main methods for generating pairwise alignments:
1. Dot-Matrix Method
The dot-matrix method, also known as the dot plot method, is a graphical approach to sequence alignment that visually compares two sequences using a two-dimensional matrix.
In this method, the two sequences to be compared are plotted along the horizontal and vertical axes of the matrix. The technique scans each residue in one sequence for similarities with all residues in the other sequence. When a residue in one sequence matches a residue in the other, a dot is placed at the corresponding position in the matrix; otherwise, that position remains blank.
If the two sequences are highly similar, the resulting dot plot will appear as a continuous line along the matrix’s main diagonal. Conversely, if the sequences have less similarity, the dot plot will exhibit a more scattered pattern of dots with fewer diagonal lines, indicating reduced similarity. Additionally, dot plots can reveal repeat elements within a single sequence, with short parallel lines above and below the main diagonal suggesting the presence of such repeats.

Figure: Example of comparing two sequences using dot plots. (Xiong, J., 2006).
2. Dynamic Programming
Dynamic programming is a powerful technique used to find the optimal alignment between two protein or nucleic acid sequences by systematically comparing all possible pairs of characters in the sequences. This method can produce both global and local alignments. The global pairwise alignment algorithm using dynamic programming is based on the Needleman-Wunsch algorithm, while the local alignment is based on the Smith-Waterman algorithm. The dynamic programming approach follows three main steps:
- Initialization of the Scoring Matrix:
The first step involves creating a two-dimensional matrix, where the two sequences to be aligned are placed along the top and left edges. This matrix is initialized with gap penalties, and the top-left corner is set to zero. - Matrix Filling with Maximum Scores:
The next step is to fill the matrix with scores according to a specified scoring system. For nucleotide sequences, scoring is straightforward: a positive score is assigned for a match, and a negative score for a mismatch. For amino acid sequences, scoring matrices like BLOSUM or PAM are utilized. The algorithm begins at the upper-left corner and moves one row at a time toward the lower-right corner, filling each cell with the maximum score obtainable by aligning the corresponding residues. - Traceback to Identify Optimal Alignment:
Once the matrix is filled, the algorithm performs a traceback to determine the optimal alignment path. Starting from the bottom-right corner and moving toward the top-left corner, it examines adjacent cells in reverse order to identify the best path that yields the highest total score. The alignment path with the maximum score represents the optimal alignment.
3. Word or k-Tuple Method
The word or k-tuple method is a heuristic approach widely recognized for its implementation in database search tools such as FASTA and BLAST. This method accelerates the alignment process by focusing on short identical sequences known as words or k-tuples.
The word method operates as follows:
- It begins by identifying these short identical sequences in the two sequences being compared.
- Once the words or k-tuples are found, dynamic programming is then employed to align the sequences based on these identified words.
This approach is notably efficient, making it suitable for rapid sequence alignment and large-scale database searches.
Methods of Multiple Sequence Alignment
Multiple Sequence Alignment Approaches
Multiple sequence alignment can be conducted using either exhaustive or heuristic approaches.
1. Exhaustive Algorithms
Exhaustive alignment involves evaluating all possible alignments simultaneously. To perform multiple sequence alignment with an exhaustive algorithm, a multidimensional search matrix is utilized, analogous to the two-dimensional matrix used in dynamic programming for pairwise alignment. This means that for aligning NN sequences, an NN-dimensional matrix is required.
While dynamic programming is a robust method for aligning sequences, it becomes computationally impractical for large datasets. As the number of sequences increases, both the computational time and memory requirements grow significantly. Consequently, exhaustive algorithms are generally limited to small datasets, typically involving fewer than ten short sequences, where the exhaustive examination of all alignments is feasible.
2. Heuristic Approaches
Heuristic approaches are employed for larger datasets to provide a more efficient alignment process. These methods prioritize speed and practicality over exhaustive accuracy, allowing for the alignment of many sequences simultaneously without the computational burden associated with exhaustive algorithms. Heuristic methods, while potentially sacrificing some alignment accuracy, are essential for handling the vast amounts of data typically encountered in biological research.
Heuristic algorithm

Figure: Progressive alignment procedure (Xiong, J., 2006)
Iterative Method
The iterative method is a dynamic approach used for multiple sequence alignment that focuses on refining an initial suboptimal solution through repeated modifications until an optimal alignment is achieved.
Steps Involved in the Iterative Method:
- Initial Pairwise Alignment:
The process begins with an initial pairwise alignment, which is used to create a guide tree. This tree assigns weights that will be utilized for subsequent alignments. - Identification of Aligned Regions:
Aligned regions containing gaps are identified in the initial alignment. These regions are then iteratively adjusted to improve the overall alignment score. - Calculation of New Alignments:
The highest-scoring alignment from the previous step is used to inform a new set of calculations. This includes predicting a new tree, recalculating weights, and generating new alignments based on the improved scores. - Iteration Until Convergence:
The procedure is repeated until no further improvement can be made in the alignment score, indicating that an optimal alignment has been reached.
An example of a web-based program that implements the iterative method is PRRN, which is specifically designed for aligning multiple sequences while iteratively refining the results to enhance alignment accuracy. This approach effectively balances computational efficiency and alignment quality, making it suitable for various bioinformatics applications.

Figure: Iterative alignment procedure for PRRN (Xiong, J., 2006)
Block-Based Method
The block-based method is a local alignment approach designed to effectively align highly divergent sequences of varying lengths, especially when traditional global alignment methods may struggle to identify conserved domains and motifs.
Key Features of the Block-Based Method:
- Identification of Shared Blocks:
This method focuses on identifying blocks of ungapped alignment that are shared across all sequences being compared. By concentrating on these conserved segments, the method enhances the accuracy of the alignment for divergent sequences. - Local Alignment Approach:
Unlike progressive and iterative methods that rely on global alignment, the block-based method employs a local alignment strategy. This allows for the alignment of regions within sequences that are similar, even if the overall sequences differ significantly. - Handling of Divergent Sequences:
The block-based method is particularly effective for aligning sequences that are evolutionarily distant or of different lengths. By targeting conserved blocks, it can reveal functional and structural similarities that may be obscured in a global alignment.
Overall, the block-based method provides a robust framework for aligning sequences with varying degrees of similarity, making it a valuable tool in bioinformatics for studying conserved motifs and domains across divergent sequences.
Applications of sequence alignment
Sequence alignment is a fundamental technique in bioinformatics that has numerous applications across various fields, including genomics, proteomics, and evolutionary biology. Below are some detailed applications of sequence alignment:
1. Identification of Unknown Sequences
One of the primary applications of sequence alignment is the identification of unknown biological sequences by comparing them to known sequences stored in databases. When a new sequence is obtained (e.g., through sequencing technologies), researchers can use alignment algorithms to match it against existing databases, such as GenBank or UniProt. By finding similar sequences, researchers can infer the function, origin, and potential role of the unknown sequence based on its alignment with known sequences.
2. Characterization of Conserved Patterns and Motifs
Sequence alignment is instrumental in identifying conserved sequence patterns and motifs, which are short, recurring sequences that have a biological significance. These conserved regions often correlate with functional domains in proteins or regulatory elements in DNA sequences. By analyzing multiple aligned sequences, researchers can determine which parts of the sequence are essential for function, allowing for better understanding of the biological roles of genes and proteins. Identifying motifs can also aid in predicting the function of newly discovered proteins.
3. Construction of Phylogenetic Trees
Phylogenetic analysis relies heavily on sequence alignment to understand evolutionary relationships among different organisms. By aligning homologous sequences from various species, researchers can assess the degree of similarity and divergence among these sequences. The resulting alignment can be used to construct phylogenetic trees, which visually represent the evolutionary history and relationships between species. This information is crucial for studies in evolutionary biology, ecology, and conservation genetics.
4. Prediction of Protein Structures
Sequence alignment plays a significant role in predicting the secondary and tertiary structures of proteins. By aligning a newly sequenced protein with homologous proteins of known structure, researchers can infer the likely structure of the target protein based on conserved regions that correlate with structural elements. Tools like SWISS-MODEL utilize sequence alignments to create homology models, which can provide insights into protein function, interaction, and dynamics.
5. Gene Location Prediction and Gene Family Identification
Sequence alignment can aid in predicting gene locations within a genome. By comparing genomic sequences with known genes, researchers can identify potential gene regions, including coding and non-coding sequences. Additionally, sequence alignment can be used to identify new members of gene families by aligning related sequences. This is particularly useful in understanding gene duplication events and the evolution of gene families, which can have significant implications in functional genomics and evolutionary studies.
6. Development of Degenerate PCR Primers
Degenerate PCR primers are designed to amplify a range of similar sequences, making sequence alignment essential in their development. By analyzing multiple related sequences, researchers can identify conserved regions suitable for primer design, which increases the likelihood of successfully amplifying target sequences that may have slight variations. This application is particularly valuable in studies of genetic diversity, evolutionary biology, and metagenomics, where multiple variants of a gene are present.
Conclusion
In summary, sequence alignment is a powerful tool with diverse applications in bioinformatics. Its ability to compare and analyze sequences enables researchers to uncover biological insights, understand evolutionary relationships, predict protein structures, identify conserved motifs, and design effective PCR primers. As sequencing technologies continue to advance, the importance of sequence alignment in biological research and its applications will only grow, paving the way for new discoveries and innovations in the life sciences.
Related posts:
![Humangenomeproject-personalizedmedicine]()
Bioinformatics in 2023: The Job Landscape Unraveled
bioinformatics![3Dstructureofprotein-deepmind]()
Frequently asked questions (FAQ's) about Alpha Fold
bioinformatics![openai-codex]()
OpenAI codex software can turn English instruction into Programming Code
A.I![variantcalling-bioinformatics]()
Step-by-Step Manual: How to Manage Your Files & Directories for Bioinformatics Projects
bioinformatics![FASTA-protein sequence-proteome.]()
Step-by-Step Guide: Convert Multiline FASTA to Single-Line FASTA
bioinformatics![data-science]()
Introducing Data Science to Undergraduate Students: A Practical Approach Using Bioinformatics
bioinformatics![bioinformatics database-mysql-php]()
Navigating the Complexities of Data Sharing in Bioinformatics
bioinformatics![A.I battle - Chatgpt, bard, claude, perplexity, Pi]()
Artificial intelligence innovation course in bioinformatics
bioinformatics![bioinformatics projects]()
Bridging Science and Technology: The Fascinating World of Bioinformatics and Beyond
bioinformatics![IT-Enabled Precision Medicine and Genomics]()
IT-Enabled Precision Medicine and Genomics
A.I![COVID-19-SARS-CoV-2-deletedsequence.]()
Bioinformatic Trends in Understanding SARS-CoV-2 Variants and Human Genomics
bioinformatics![A-RNA-sequence-analysis-basics.]()
Step-by-Step Guide: Make your first bioinformatics project
bioinformatics![AI-bioinformatics]()
Mastering Bioinformatics in 2025: Essential Skills and Emerging Trends
bioinformatics![Bioinformatics]()
Guide to Installing, Running, and Analyzing MD Simulations with GROMACS: A Practical Approach with P...
bioinformatics![moleculargenetics]()
Tackling Bias in Gene Expression Dataset Selection
A.I![bioinformatics]()
The Definitive Guide to Bioinformatics Tools, Software, and Databases
bioinformatics